La lettre Surrénale – Mai 2013
 |
||||||||
|
||||||||
|
||||||||
|
La corticosurrénale : de la cellule au patient Bienvenue à LS2 ! Cette deuxième lettre est consacrée exclusivement à la corticosurrénale, vue sous des angles bien différents : du côté des sciences fondamentales et translationnelles, Antoine Martinez nous livre, dans une saga “déconstruite”, les secrets d’une zone fondamentale à l’unité fœtoplacentaire alors qu’Estelle Louiset décortique les mécanismes moléculaires de l’hyperaldostéronisme primaire. Plus près des cliniciens, votre serviteur vous dévoile les nouveaux vêtements d’un vieux médicament et Laurence Guignat vous fera prendre conscience de la différence vitale entre savoir, faire savoir et savoir-faire. Enfin, au plus près des malades, Claudine Colin vous présentera leur remarquable association. Volontairement nous avons présenté cette liste à l’envers : priorité aux patients ! Bonne lecture et rendez-vous à la LS3, où l’on parlera aussi médullaire et phéochromocytome… |
||||||||
 |
||||||||
|
Association Surrénales L’association Surrénales (www.surrenales.com) a été créée le 12 septembre 1996 et déclarée d’intérêt général le 11 mai 2006. Au 15 avril 2013, 644 adhérents, dont 556 concernés par les maladies des surrénales, sont répartis dans 5 entités nouvellement mises en place : • Hyperplasie (HCS) et hypoplasie congénitales des surrénales : 151 • Maladie d’Addison : 147 • Syndrome de Cushing et corticosurrénalome : 116 • Insuffisance corticotrope : 83 • Syndrome de Conn et phéochromocytomes : 59 Objectifs • Informer sur les maladies des surrénales • Rompre l’isolement des malades et des familles • Soutenir la recherche Moyens • Des documents, plaquettes et livrets d’information rédigés en collaboration avec les experts des centres de référence et/ou de compétence (maladie d’Addison, syndrome de Cushing, insuffisance corticotrope, hyperplasie congénitale des surrénales, phéochromocytomes et paragangliomes), ainsi qu’un guide pratique sur le bon usage du mitotane dans les maladies de la corticosurrénale. • Des réunions organisées dans toute la France dans les centres de compétence qui rassemblent les patients, les endocrinologues et les pédiatres endocrinologues. • Un site Internet (www.surrenales.com) régulièrement mis à jour et très visité (2 500 personnes en moyenne par semaine). C’est un relais pour la diffusion de l’information, très utile notamment lors de problèmes de disponibilité des médicaments, comme récemment pour le kétoconazole ou la fludrocortisone. Recherche • Grâce aux dons de ses adhérents, l’association a pu soutenir des projets de recherche menés dans plusieurs centres de référence et de compétence sur les différentes maladies des surrénales. • L’association a maintenant un partenaire privilégié pour l’HCS : l’IFCAH (International Fund for research on Congenital Adrenal Hyperplasia) [www.ifcah.org]. Ce fonds de dotation a pour but d’apporter un soutien de grande ampleur aux recherches les plus prometteuses sur l’hyperplasie congénitale des surrénales. Conseil scientifique Depuis 2011, l’association s’est dotée d’un conseil scientifique qui se réunit 1 fois par an lors du congrès annuel de la SFE. |
||||||||
 |
||||||||
|
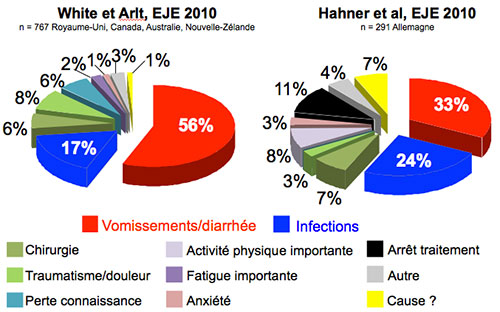
Éducation thérapeutique des patients insuffisants surrénaliens : un outil encore insuffisamment utilisé pour éviter ou traiter précocement l’insuffisance surrénale aiguë Insuffisance surrénale aiguë : une complication rare, méconnue, engageant le pronostic vital L’insuffisance surrénale aiguë est une urgence thérapeutique qui met en jeu le pronostic vital. Les sites d’associations de patients se font l’écho de mésaventures de patients pour lesquels la gestion de la crise d’insuffisance surrénale n’a non seulement pas été optimale, mais a même entraîné des décès attribuables à une décompensation aiguë (http://www.addisons.org.uk/info/experiences). Une insuffisance surrénale aiguë a été retenue comme étant responsable de 36 des 507 décès (7 %) du registre suédois de patients addisonniens (1) et de 59 des 106 décès brutaux inattendus (56 %) dans une cohorte américaine de 6 107 patients traités par hormone de croissance (2). Son incidence est d’environ 6 à 7,5/100 patients-année (3, 4). Entre 20 et 44 % des patients (traités) ont eu au moins 1 crise d’insuffisance surrénale aiguë et 10 % des patients ont déjà fait plusieurs crises aiguës (3-7). Il n’existe pas de profil type du patient susceptible de faire une insuffisance surrénale aiguë, même si certaines facteurs ont été retrouvés associés à un risque accru de décompensation, comme l’origine périphérique de l’insuffisance surrénale, le sexe féminin, l’âge élevé lors du diagnostic d’insuffisance surrénale, ou encore des troubles des fonctions supérieures ou un diabète insipide (4, 7, 8). Les principaux facteurs de décompensation (ou les premiers symptômes) sont les troubles digestifs (vomissements et/ou diarrhée) dans 33 à 56 % des cas et les infections dans 17 à 24 % des cas (3, 4) [figure].
Légende. Fréquence des différents facteurs de décompensation d’insuffisance surrénale aiguë dans 2 grandes études.
Les autres facteurs sont multiples : une chirurgie, un accouchement, une forte émotion, des douleurs intenses, une fatigue importante, une interruption du traitement, etc. Parfois, aucun facteur de décompensation n’est identifiable. Éducation thérapeutique du patient insuffisant surrénalien : une nécessité Le manque de spécificité des signes d’insuffisance surrénale aiguë et la méconnaissance de cette urgence médicale rare sont susceptibles de conduire à un retard de mise en route du traitement ou à des examens invasifs pouvant précipiter une issue fatale. Un certain nombre d’outils existent depuis de nombreuses années, comme la carte d’insuffisance surrénale ou d’autres dispositifs d’alerte, la prescription d’hydrocortisone injectable, la diffusion d’informations aux patients et l’établissement de protocoles d’urgence. Si la plupart des patients possèdent une carte d’urgence, trop peu disposent d’hydrocortisone injectable (3, 4). Le niveau des connaissances sur l’insuffisance surrénale aiguë, évalué au moyen d’autoquestionnaires, était très bon chez seulement 28 % des 25 patients d’une étude anglaise (5) et chez 5 % des 84 patients d’une étude danoise (9) dans les années 1990. Plus inquiétant encore, parmi les patients qui se considéraient comme étant bien informés sur l’insuffisance surrénale, 42 % prenaient de mauvaises décisions en mise en situation (9). Enfin, une étude canadienne révèle que, sur une période allant de 1998 à 2007, moins de la moitié des parents d’enfants admis en hospitalisation pour une décompensation surrénalienne aiguë avaient bien augmenté l’hydrocortisone avant de se rendre aux urgences, et seuls deux tiers des urgentistes et autres médecins hospitaliers appliquaient le protocole d’urgence (10). Chez les patients asthmatiques, l’inefficacité des approches éducatives limitées à la transmission d’information a bien été démontrée (11), de même que l’absence d’autoapprentissage après une décompensation aiguë (12). Il paraît donc essentiel d’aider les patients à acquérir ou à maintenir les compétences dont ils ont besoin pour gérer au mieux leur vie avec une maladie chronique, ce qui est l’objectif de l’éducation thérapeutique. Plusieurs programmes d’éducation thérapeutique couvrant le champ de l’insuffisance surrénale ont reçu l’autorisation des Agences régionales de santé (ARS), notamment dans les hôpitaux parisiens Cochin, Robert-Debré et Pitié-Salpêtrière, ainsi qu’à l’hôpital de la Timone, à Marseille. Les thématiques communes sont la gestion quotidienne du traitement substitutif, l’adaptation des doses d’hydrocortisone aux facteurs intercurrents et l’auto-injection intramusculaire d’hydrocortisone. Les entretiens permettent d’appréhender les besoins et les attentes du patient, ses représentations et ses croyances en matière de santé, ses difficultés et ses atouts. Plusieurs difficultés ont souvent été rencontrées : les difficultés liées au patient, telles les “corticophobies”, le manque de confiance en ses connaissances, la crainte de mal faire, la croyance en la toute-puissance de la parole médicale, etc., mais aussi les difficultés liées à l’environnement, comme l’activité professionnelle en extérieur, l’exposition aux infections, les voyages fréquents à l’étranger, etc. Parmi les outils utilisés, on peut citer : les courbes prédécoupées de doses d’hydrocortisone à placer sur une frise temporelle ; les cartes des situations à risque ; le jeu des comprimés d’hydrocortisone à placer sur une frise temporelle ; l’intervention d’un patient expérimenté, etc. Alors que les diabétologues jonglent depuis de nombreuses années avec les méthodes pédagogiques actives, les endocrinologues doivent s’approprier ces techniques pour les appliquer à d’autres maladies chroniques afin de faciliter les apprentissages. Depuis l’inscription de l’éducation thérapeutique du patient dans la loi Hôpital, patients, santé et territoires du 21 juillet 2009, les programmes d’éducation thérapeutique du patient doivent répondre au cahier des charges national et sont soumis à l’autorisation des ARS. C’est une opportunité à saisir pour améliorer l’organisation et la qualité de la prise en charge de nos patients, même si, comme souvent, le financement de cette activité reste flou.
Références bibliographiques |
||||||||
|
…………………………………………………………………………………………………………………………………………………………… De nouvelles formes galéniques d’hydrocortisone pour le traitement substitutif de l’insuffisance surrénale Le traitement substitutif actuel de la carence en cortisol de l’insuffisance surrénale est loin d’être idéal. Le choix de la molécule ne pose pas de problèmes : la plupart des patients sont traités par de l’hydrocortisone, qui n’est qu’un autre nom du cortisol, même si 25 % reçoivent d’autres agonistes du récepteur glucocorticoïde (1). Une des difficultés réside dans la cinétique de sécrétion physiologique du cortisol, le fameux “cycle nycthéméral”, au cours duquel la sécrétion de cortisol est quasiment nulle vers minuit, démarre en deuxième partie de nuit pour réaliser un pic de sécrétion le matin au lever, puis décroît dans la journée, avec une discrète inflexion aux repas, pour redevenir très basse au coucher (2). Ce cycle est impossible à reproduire avec la forme galénique actuelle de l’hydrocortisone (comprimé de 10 mg avec une demi-vie courte), ce qui pose 2 problèmes. Le premier problème est que, pour assurer une bonne “couverture” en cortisol dans la journée, il faut idéalement 3 doses d’hydrocortisone, en quantité décroissante selon un schéma 1 ; 1/2 ; 1/4, doses prises avant les repas et adaptées au poids : par exemple, pour un patient de 75 à 84 kg, on donnera 10 mg le matin, 5 mg le midi et 2,5 mg le soir (2). Ce schéma n’est pas facile à respecter par le patient, qui doit couper des comprimés en 4 et ne pas oublier la prise à midi. Le deuxième problème est que le matin, au lever, le taux de cortisol est très bas, alors qu’il devrait au contraire être le plus élevé. Cela est sans doute un des facteurs qui contribuent à la mauvaise qualité de vie des patients insuffisants surrénaliens (3). Bien évidemment, on ne peut pas résoudre ce problème de cortisol très bas le matin par une plus forte prise d’hydrocortisone (ou d’un autre glucocorticoïde) le soir au coucher, car on soumettrait alors nécessairement le patient à une forte exposition au cortisol la nuit, ce qui est responsable d’effets métaboliques délétères. Actuellement, la seule façon de reproduire le cycle nycthéméral de cortisol consiste à mettre en place une pompe sous-cutanée à débit variable, comme pour l’insuline dans le diabète de type 1, ce qui est possible, mais trop lourd. Pour essayer d’améliorer le traitement substitutif oral par hydrocortisone, 2 approches sont developpées actuellement, qui concernent toutes 2 la galénique. La première approche, développée par Richard J Ross, vise à trouver une galénique de l’hydrocortisone permettant une libération différée. Le patient la prendrait le soir au coucher, et elle commencerait à se libérer au milieu de la nuit pour donner un taux maximal au lever. Cette approche séduisante, qui a conduit au développement de comprimés contenant de l’hydrocortisone au centre et un composé inerte en périphérie, a déjà été expérimentée dans le traitement substitutif de l’insuffisance surrénale (4) et de l’hyperplasie congénitale des surrénales (5), et elle donne des résultats prometteurs. Elle se heurte cependant à des problèmes d’industrialisation, la forme galénique actuellement étudiée étant des microparticules qui semblent prometteuses (RJ Ross, communication orale, Journées de l’hyperplasie congénitale des surrénales, Paris, 5 avril 2013). La deuxième approche, qui, elle, a franchi le cap de la production industrielle et devrait prochainement être disponible, a consisté à développer un comprimé à libération double, que nous appellerons CLD: la partie externe du comprimé est à libération rapide et la partie centrale à libération plus lente. Avec ce type de comprimé, le patient se réveille avec un taux de cortisol aussi bas qu’avec un traitement classique, mais il prend dès le lever le CLD qui vise 2 objectifs : une ascension rapide des taux de cortisol le matin et des taux soutenus plus longtemps pendant le reste de la journée. Une étude suédoise utilisant ce type de comprimé a été publiée l’année dernière dans le Journal of Clinical Endocrinology and Metabolism (6). Dans cette étude prospective randomisée ouverte avec cross over portant sur 64 patients insuffisants surrénaliens primaires (53 non diabétiques et 11 diabétiques), Le CLD en 1 prise matinale a été comparé, à quantité journalière d’hydrocortisone égale, aux 3 prises quotidiennes de comprimés d’hydrocortisone classiques, pendant 2 périodes de 12 semaines. Les objectifs étaient de comparer les profils pharmacocinétiques du cortisol plasmatique et l’évolution de paramètres pouvant être perturbés par une trop forte exposition au cortisol, notamment le poids, la tension artérielle, l’hémoglobine glyquée. Il est important de noter que dans cette étude, lorsqu’ils étaient dans le bras “3 prises d’hydrocortisone classique”, les patients recevaient une dose journalière totale d’hydrocortisone relativement élevée. Il s’agissait en fait de leur dose habituelle avant l’étude mais celle-ci était curieusement bien au-dessus de la dose proposée par P.M. Mah et al. (2). En effet, le poids moyen de ces 64 patients insuffisants surrénaliens étant de 79,6 kg, ils auraient théoriquement dû recevoir une posologie journalière de 17,5 mg d’après le schéma de référence (2) alors qu’ils recevaient 30 mg/j.
• L’aire sous la courbe de cortisolémie sur 24 heures obtenue par le schéma “1 prise CLD” ne représente que 77 % de celle du schéma de référence “3 prises hydrocortisone” à la même posologie journalière. Cela veut dire que, en termes d’exposition au cortisol, 10 mg de CLD sont équivalents à 7,7 mg d’hydrocortisone classique (en 3 prises). Une autre façon de présenter ce résultat est de dire qu’il faudrait augmenter la posologie en hydrocortisone de 30 % du CLD pour obtenir une exposition au cortisol équivalente à celle donnée par les 3 comprimés d’hydrocortisone classique : ainsi, pour obtenir une exposition équivalente à 10 mg d’hydrocortisone (en 3 prises) par un CLD, il faudrait un CLD dosé à 13 mg (10 : 7,7 × 10) d’hydrocortisone. • Le taux maximal du cortisol obtenu par “1 prise CLD” est un peu inférieur (691 nmol/l versus 803) et son délai de survenue est un peu plus tardif (67 minutes versus 50). En revanche, l’aire sous la courbe des 4 premières heures après la prise est un peu supérieure (de 6,5 %). Cela montre que, malgré un pic un peu inférieur et un peu plus tardif, le CLD est capable d’assurer une imprégnation en cortisol un peu plus importante que l’hydrocortisone classique pendant les 4 premières heures. • Les aires sous la courbe des intervalles 4 à 10 heures après la prise, et 10 à 24 heures après la prise, sont en revanche inférieures de 28 et de 56 % dans le groupe “1 prise CLD” par rapport au schéma “3 prises d’hydrocortisone” pris en référence.
Les périodes de traitement “1 prise CLD” montrent une évolution (valeurs sous CLD ? valeurs sous hydrocortisone 3 prises) significative du poids (? 0,7 kg), des tensions systolique (? 5,5 mmHg) et diastolique (? 2,3 mmHg), et de l’HbA1c (? 0,1, et, chez les 11 patients diabétiques, ? 0,6). Ces évolutions vont donc toutes dans le sens d’une évolution favorable de ces paramètres sous CLD. Au total, dans cette étude où il est comparé à un schéma d’hydrocortisone en 3 prises à posologie équivalente, le CLD en 1 seule prise montre une biodisponibilité globale inférieure de 23 %, probablement liée à une moindre absorption digestive, avec des taux de cortisol plasmatique un peu supérieurs dans les 4 premières heures, qui deviennent plus bas ensuite. De plus, le CLD est associé à une évolution favorable de paramètres métaboliques cardiovasculaires. Il est bon de souligner plusieurs points • Cette étude ne permet pas de savoir si l’effet métabolique favorable du CLD est lié à son profil pharmacocinétique différent ou à sa biodisponibilité inférieure, d’autant plus que le traitement comparatif est donné à une posologie élevée. Pour déterminer la part revenant à la pharmacocinétique, il faudrait faire une nouvelle étude dans laquelle les 2 bras auraient une exposition globale identique au cortisol, ce qui demanderait de donner des doses de CLD 30 % plus élevées que la dose d’hydrocortisone classique en 3 prises, ou de garder la même dose de CLD mais de baisser alors la dose d’hydrocortisone classique de 23 %. • Le traitement de référence de l’étude n’est pas si facile à appliquer par les patients, en raison des 3 prises et de la nécessité de couper les comprimés en 2 ou même en 4 : dans la vie réelle (expérience personnelle non publiée), il semble que beaucoup de patients ne prennent que 2 prises (voire 1 seule le matin !), ce qui limite peut-être la valeur des différences de pharmacocinétique rapportées dans cette étude. • Enfin, nous n’avons pas évoqué la question de l’adaptation indispensable du traitement pendant les périodes de stress, afin de prévenir l’insuffisance surrénale aiguë (voir l’excellent article de Laurence Guignat dans cette Lettre). Dans l’étude de G. Johannson et al. (6), les patients avaient comme consignes, pendant les situations de stress, de doubler les doses dans le schéma 3 prises et de prendre une deuxième dose de CLD 8 heures après la première dans l’autre bras. Il y a eu 6 hospitalisations pour causes infectieuses au cours de l’étude dans le bras CLD, dont 2 avec un diagnostic d’insuffisance surrénale aiguë retenue et 2 hospitalisations dans le groupe témoin pour gastroentérite, mais sans que le diagnostic d’insuffisance surrénale aiguë ait été retenu. Ces différences ne sont sans doute pas significatives, d’autant plus que l’étude était ouverte et que les décisions d’hospitalisation ont pu être biaisées par ce fait, mais elles rappellent qu’il est important de rester vigilant sur le risque d’insuffisance surrénale aiguë après tout changement thérapeutique chez un patient insuffisant surrénalien. La mortalité des patients insuffisants surrénaliens est 2 fois plus élevée que la mortalité attendue et une partie de cette surmortalité est liée à des insuffisances surrénales aiguës mal gérées. Malgré ces interrogations, il faut souligner que la prise unique qu’offre le CLD représente un avantage important pour les patients, en raison précisément de leur difficulté à prendre l’hydrocortisone en 3 ou même en 2 prises. Dans l’étude de G. Johannsson et al., 85 % des patients ont exprimé une préférence grande ou très grande pour le CLD et 92 % des patients ont choisi de continuer dans l’étude d’extension (6). Au final, il faut saluer une nouveauté dans le traitement de l’insuffisance surrénale, et de nouvelles études viendront sans doute préciser l’intérêt des nouvelles formes galéniques de l’hydrocortisone, existantes et à venir.
Références bibliographiques |
||||||||
|
…………………………………………………………………………………………………………………………………………………………… Panne de pompes dans la surrénale : mutations de Na+/K+ et Ca2+ ATPases dans les adénomes de Conn L’hyperaldostéronisme primaire (HAP), qui concerne 6 à 10 % des patients hypertendus, est une cause endocrinienne classique d’hypertension, suspectée devant l’association hypertension artérielle + hypokaliémie + hyporéninémie. Différentes pathologies corticosurrénaliennes sont responsables d’HAP, l’adénome de Conn représente approximativement 50 % des cas, alors que les hyperplasies unilatérale et bilatérale des zones glomérulées touchent respectivement 10 et 40 % des patients. La connaissance des processus moléculaires aboutissant au développement des différentes formes d’HAP est très limitée. À ce jour, aucune altération de région chromosomique n’a pu être associée au développement des adénomes de Conn par hybridation génomique comparative (Comparative Genomic Hybridization [CGH]) [1]. Des mutations germinales et somatiques affectant le gène codant le canal potassique KCNJ5 ont été rapportées dans des cas familiaux et sporadiques d’HAP, en proportion variable selon les séries (2-4). Dans le numéro d’avril 2013 de la revue Nature Genetics sont décrites pour la première fois des mutations somatiques des gènes codant les pompes Na+/K+ ATPase (ATP1A1, NM_000701.7) et la Ca2+ ATPase (ATP2B3, NM_021949.3) dans des adénomes de Conn (5). La pompe Na+/K+ ATPase est connue pour contribuer au maintien du potentiel de repos des cellules en permettant l’efflux de 3 Na+, en contrepartie d’un influx de 2 K+. La pompe Ca2+ ATPase, qui est capable d’extraire les ions Ca2+ des cellules, participe au contrôle de l’homéostasie calcique. Pour mener à bien ce travail, les équipes du réseau européen ENS@T (avec une forte participation du réseau français INCa-COMETE) ont réalisé le séquençage de l’exome du tissu surrénalien tumoral de 9 patients porteurs d’HAP qui ne présentaient pas de mutation KCNJ5. L’analyse des exomes a révélé la présence de mutations affectant ces ATPases dans 3 cas. Le séquençage des 2 gènes, réalisé sur une série de plus de 300 tissus de la collection biologique du réseau ENS@T, a permis d’identifier plusieurs mutations somatiques chez 21 sujets (6,8 %). Aucune mutation germinale des gènes ATP1A1 et ATP2B3 n’a été détectée chez ces patients, ni chez les 18 cas d’hyperaldostéronisme familial, ni dans les 91 cas d’hyperplasie bilatérale associée à un HAP examinés dans cette étude. Pour le gène ATP1A1, les atteintes observées dans les adénomes de Conn correspondaient à des mutations ponctuelles (c.311T>G, c.995T>G et c.1738A>G) entraînant des substitutions Leu104Arg, Val332Gly et Ile580Val, ainsi qu’une délétion (c.299_313delTCTCAATGTTACTGT) responsable d’une perte de la région Phe100_Leu104. Les études fonctionnelles entreprises sur des modèles cellulaires (cellules COS et HEK) exprimant le gène ATP1A1 altéré ont montré que les mutations ponctuelles sont responsables d’une baisse de liaison du K+ et d’une perte d’activité enzymatique de la protéine qui entraînent une dépolarisation des cellules. Pour le gène ATP2B3, ont été observées des délétions (c.1272_1277delGCTGGT, c.1273_1278delCTGGTC et c.1277_1282delTCGTGG) à l’origine de la perte de 2 acides aminés 425-426 ou 426-427 (p.Leu425_Val426del ou p.Val426_Val427del) de la protéine dans une région située à proximité du site de liaison des ions Ca2+. Les études électrophysiologiques ont révélé que les cellules corticosurrénaliennes tumorales des patients porteurs d’une mutation affectant le gène ATP1A1 ou ATP2B3 présentaient une moindre polarisation membranaire que les cellules péritumorales. Sur la base de ces données, les chercheurs concluent que, dans les adénomes de Conn, les mutations inactivatrices de ATP1A1 sont responsables d’une dépolarisation cellulaire qui facilite l’ouverture des canaux calciques voltage-dépendants.
Références bibliographiques
|
||||||||
|
…………………………………………………………………………………………………………………………………………………………… Zone fœtale de la corticosurrénale : saga génétique d’une zone en transit Des modèles génétiques murins précisent les mécanismes conduisant à la persistance de la zone fœtale (SUMOylation de Sf-1 ou activation de la PKA) ou au contraire à son absence (déficit du facteur GATA-6).
Chez le fœtus de l’homme et des primates supérieurs, la glande corticosurrénale est très développée. Elle se compose d’un néocortex périphérique (ou zone définitive), hormonalement très peu actif, qui sera à l’origine du cortex adulte et d’une énorme zone spécialisée occupant 80 à 90 % du cortex, appelée la zone fœtale, qui concentre une extraordinaire capacité stéroïdogène (1). Dans la glande surrénale de souris, la zone X est considérée comme l’équivalent de la zone fœtale humaine. Jusqu’à récemment, cette parenté était très discutée, car les observations anatomiques et fonctionnelles avaient certes fourni des éléments convergents, mais d’autres restaient contradictoires. Chez les 2 espèces, ces zones corticales partagent une même position juxtamédullaire et leur présence est transitoire, puisqu’elles régressent progressivement pour disparaître de l’organe adulte. Cependant, contrairement à la zone fœtale humaine qui produit de grandes quantités de DHEA et DHEAS, précurseurs obligatoires des estrogènes placentaires essentiels au maintien de la gestation, la zone X n’a pas d’activité stéroïdogène connue. Enfin, la régression de la zone fœtale humaine commence à la naissance pour s’achever généralement vers 3 mois, alors que chez la souris, la régression de la zone X est différée à la maturité sexuelle. L’origine fœtale de la zone X est maintenant bien établie. La démonstration chez la souris que les cortex définitif (adulte) et transitoire (fœtal) dérivent d’un même primordium embryonnaire exprimant le gène Sf-1, puis suivent des programmes de différenciation différents, a été apportée en 2008 par des expériences de lignage cellulaire réalisées dans le laboratoire de K.I. Morohashi et al. (2). La découverte des mécanismes transcriptionnels responsables de l’expression surrénalienne du gène Sf-1 a permis de tracer 2 populations d’adrénocytes embryonnaires, qui diffèrent par les séquences cis-régulatrices mises en jeu pour maintenir la transcription de Sf-1 : les cellules engagées dans un programme de différenciation fœtale (future zone X) maintiennent active une séquence enhancer “fœtale” présente dans l’intron 4 de Sf-1, tandis que celles engagées dans un programme de différenciation adulte (futur cortex définitif) répriment cette séquence fœtale tout en conservant l’expression de Sf-1 par la mobilisation d’une séquence enhancer “adulte” qui reste à identifier. Cependant, l’histoire ne disait pas si Sf-1 était le levier ou le témoin de cette programmation, ni comment 2 populations cellulaires de même origine pouvaient suivre un destin différent en exprimant le même facteur. Dans les 3 épisodes qui suivent, nous allons présenter des éléments de réponses à ces questions, et nous nous permettons de raconter ces épisodes dans un ordre qui n’est pas chronologique : commençons par le deuxième.
H.A. Ingraham et al. ont apporté une réponse inattendue à cette question en s’intéressant à la SUMOylation de Sf-1 (3). La conjugaison de peptides SUMO (Small Ubiquitin-like Modifier) sur le facteur Sf-1 est une modification post-traductionnelle réversible qui réprime son activité transcriptionnelle en affectant sa liaison à l’ADN. Cependant, dans les promoteurs des gènes cibles de Sf-1, cette perte d’interaction ne concerne que des sites à faible affinité “non consensus”, qualifiés alors de SUMO-sensibles, tandis que la liaison aux sites consensus SUMO-résistants reste, elle, inchangée (4). Ce concept de gènes SUMO-sensibles suggère qu’en contrôlant les cycles de SUMOylation/déSUMOylation d’un seul facteur, il est possible d’activer différentes combinaisons de gènes cibles et donc de déclencher différents programmes génétiques. L’équipe de H.A. Ingraham est allée plus loin en testant ce concept in vivo en remplaçant le gène Sf-1 des souris par un gène mutant codant un Sf-1 rendu inSUMOylable par la substitution des lysines K119 et K194 en arginine (souris Sf-12KR) [3]. Les souris Sf-12KR sont le premier modèle génétique mammifère permettant d’explorer les conséquences de la SUMOylation d’un substrat endogène. Ces souris présentent des altérations du programme de développement adrénogonadique avec une expression de marqueurs corticosurrénaliens dans le testicule (Shh, Cyp21a1) et d’un marqueur testiculaire (Sox9) dans la surrénale, et enfin une persistance de la zone fœtale dans le cortex adulte. Les auteurs expliquent ces défauts développementaux par le “réveil” de gènes SUMO-sensibles présentant dans leur région cis-régulatrice des sites de faible affinité (non consensus) inaccessibles à la forme SUMOylée de Sf-1 mais qui seront spécifiquement activables par la forme inSUMOylable du facteur ; ils le montrent formellement pour le gène Shh et le postulent pour les autres. Ces travaux démontrent de manière indiscutable l’effet répresseur de la SUMOylation de Sf-1, mais, surtout, suggèrent que le contrôle de la déSUMOylation au cours de la vie embryonnaire va permettre d’activer des gènes SUMO-sensibles et modifier les programmes de développement. Ce dernier point est d’autant plus plausible que les auteurs mettent en évidence les effets dominants de l’allèle Sf-12KR qui vont s’imposer même en présence de l’allèle Sf-1 sauvage à l’état hétérozygote. Cependant, aucune explication moléculaire ne sous-tend la persistance de la zone fœtale. Venons donc à l’épisode 1.
Il existe à ce jour un seul autre modèle génétique affichant une zone fœtale persistante : les souris AdKO (5, 6). Dans ce modèle reproduisant l’hyperplasie micronodulaire corticosurrénalienne (PPNAD) observée dans le complexe de Carney, le gène Prkar1a codant la sous-unité régulatrice R1? de la PKA a été invalidé. Le déficit surrénalien de R1? induit une activation constitutive de la PKA conduisant à un syndrome de Cushing et à l’expansion d’un cortex “fœtal”. La convergence des phénotypes “fœtaux” des souris Sf-12KR et AdKO est pour le moins troublante et suggère qu’il existe un lien fonctionnel entre SUMOylation et PKA. Plusieurs travaux révèlent un rôle inhibiteur de la signalisation PKA sur la SUMOylation des protéines dans l’ovaire ou dans le tissu adipeux (7, 8). Une hypothèse vraisemblable est que l’activité PKA participe à la régulation des cycles de SUMOylation/déSUMOylation qui vont, in fine, ajuster la proportion de Sf-1 SUMOylé dans la surrénale en développement. En accord avec cette possibilité, l’équipe de H.A. Ingraham a démontré en 2008 que le gène Inha, codant l’inhibine ?, est un gène SUMO-sensible puisque son promoteur ne peut être activé par Sf-1 si celui-ci est SUMOylé (4). Or, les patients souffrant d’un PPNAD, comme les souris AdKO, présentent une surexpression de l’inhibine ? dans leurs lésions surrénaliennes. L’inhibine ? est non seulement un marqueur fœtal mais aussi un puissant inhibiteur de l’activine dont la fonction est d’induire la régression apoptotique du cortex fœtal. Il est alors possible d’imaginer que le maintien de la zone fœtale chez les souris Sf-12KR résulte du “réveil” du gène Inha. Il reste maintenant à établir chez les patients Carney souffrant de PPNAD et dans le modèle murin correspondant (AdKO) que les lésions surrénaliennes se développent dans un contexte d’hypoSUMOylation. Mais en attendant, voici l’épisode 3.
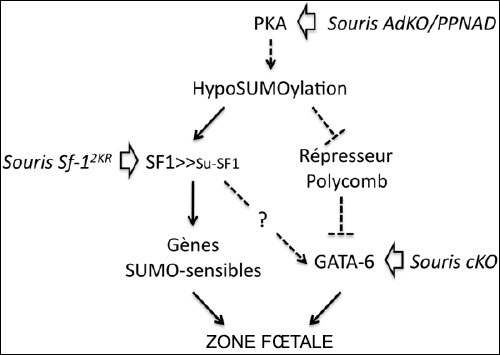
Il manquait à cette histoire une version en miroir, c’est-à-dire une situation génétique où cette fois la zone fœtale viendrait à manquer. C’est chose faite désormais puisque D.B. Wilson et al. viennent de publier un modèle de souris déficientes pour le facteur de transcription GATA-6 dans le cortex surrénal (souris cKO). Leur phénotype principal est une absence de développement de la zone fœtale sans atteinte significative du cortex définitif ni de la fonction endocrine (9). Si ces travaux montrent pour la première fois l’importance de GATA-6 dans la programmation de la zone fœtale, les auteurs ne donnent pas d’explication moléculaire à ce phénotype. Peut-on, là aussi, tenter de relier ce défaut développemental à la SUMOylation ? Cette spéculation repose sur le fait que l’hyperSUMOylation générale observée dans des souris déficientes pour la déSUMOylase SENP2 (souris Senp2-/-) conduit à un arrêt du développement embryonnaire cardiaque au stade E10, dû à la répression des gènes codant GATA-4 et GATA-6. Cette répression est la conséquence directe de l’hyperSUMOylation qui favorise le recrutement du complexe répresseur Polycomb sur les promoteurs des gènes Gata (8). On peut donc faire l’hypothèse qu’à l’inverse, il existerait un lien entre un état d’hypoSUMOylation et le développement du cortex fœtal, par l’intermédiaire de GATA-6. Finalement, on peut construire un modèle où les effets de la PKA sur la SUMOylation contrôleraient le développement de la zone fœtale en agissant à la fois sur la signalisation de Sf-1 et sur l’expression de GATA-6 (figure). Des recherches sont en cours dans notre équipe pour tester ce modèle.
Figure. Mécanismes génétiques avérés (traits pleins) ou hypothétiques (pointillés) reliant la régulation de la SUMOylation et le contrôle génétique du développement de la zone fœtale corticosurrénalienne chez la souris. Les modèles génétiques murins utilisés dans ces études et, le cas échéant, les pathologies humaines associées, sont mentionnés en italique. La possibilité que Gata-6 soit un gène cible SUMO-sensible de Sf-1 est une hypothèse plausible.
Références bibliographiques |
||||||||