La lettre surrénale de Juin 2017
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
De toutes les couleurs Amis de la surrénale, cette 8e newsletter se veut arc-en-ciel, choisissez votre pathologie : Alors, bien sûr, il manque beaucoup de couleurs : suite aux prochains épisodes qui, eux, c’est promis,
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Insuffisance surrénale (corticotrope) liée aux morphiniques : Tous les endocrinologues connaissent l’effet inhibiteur des glucorticoïdes sur la sécrétion d’adrénocorticotropine (ACTH) et de cortisol, car cet effet, facile à comprendre, est à l’origine d’une cause fréquente d’insuffisance surrénale, d’origine corticotrope, qui survient après l’arrêt d’un traitement glucocorticoïde : l’insuffisance surrénale postcortisonique, qui peut avoir un retentissement clinique indiscutable. Nous vous livrons ci-dessous une courte synthèse des réponses que nous avons trouvées dans la littérature, qui reste pauvre, sur les insuffisances corticotropes liées aux morphiniques. Mécanisme d’action des morphiniques sur l’axe corticotrope Études cliniques systématiques Insuffisance surrénale aiguë Conclusion |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Références bibliographiques 1. McDonald RK, Evans FT, Weise VK et al. Effect of morphine and nalorphine on plasma hydrocortisone levels in man. J Pharmacol Exp Ther 1959;125(3):241-7. 2. Vuong C, Van Uum SH, O’Dell LE et al. The effects of opioids and opioid analogs on animal and human endocrine systems. Endocr Rev 2010;31(1):98-132. 3. Tsagarakis S, Navarra P, Rees LH et al. Morphine directly modulates the release of stimulated corticotrophin-releasing factor-41 from rat hypothalamus in vitro. Endocrinology 1989;124(5):2330-5. 4. Grossman A, Moult PJ, Cunnah D et al. Different opioid mechanisms are involved in the modulation of ACTH and gonadotrophin release in man. Neuroendocrinology 1986;42(4):357-60. 5. Rittmaster RS, Cutler GB Jr, Sobel DO et al. Morphine inhibits the pituitary-adrenal response to ovine corticotropin-releasing hormone in normal subjects. J Clin Endocrinol Metab 1985;60(5):891-5. 6. Allolio B, Schulte HM, Deuss U et al. Effect of oral morphine and naloxone on pituitary-adrenal response in man induced by human corticotropin-releasing hormone. Acta Endocrinol (Copenh) 1987;114(4):509-14. 7. Abs R, Verhelst J, Maeyaert J et al. Endocrine consequences of long-term intrathecal administration of opioids. J Clin Endocrinol Metab 2000;85(6):2215-22. 8. Palm S, Moenig H, Maier C. Effects of oral treatment with sustained release morphine tablets on hypothalamic-pituitary-adrenal axis. Methods Find Exp Clin Pharmacol 1997;19(4):269-73. 9. Müssig K, Knaus-Dittmann D, Schmidt H et al. Secondary adrenal failure and secondary amenorrhoea following hydromorphone treatment. Clin Endocrinol (Oxf) 2007;66(4):604-5. 10. Oltmanns KM, Fehm HL, Peters A. Chronic fentanyl application induces adrenocortical insufficiency. J Intern Med 2005;257(5):478-80. 11. Pullan PT, Watson FE, Seow SS et al. Methadone-induced hypoadrenalism. Lancet 1983;1(8326 Pt 1):714. 12. Policola C, Stokes V, Karavitaki N et al. Adrenal insufficiency in acute oral opiate therapy. Endocrinol Diabetes Metab Case Rep 2014;2014:130071. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Traitement adjuvant du corticosurrénalome par mitotane : il faut absolument inclure vos patients à risque intermédiaire dans ADIUVO ! Le corticosurrénalome, que l’on doit maintenant appeler “cancer corticosurrénalien” (CCS, ou ACC en anglais) est un cancer rare et de mauvais pronostic : tous stades confondus, la survie à 5 ans n’est que de 16 à 38 % (1). Son traitement est chirurgical : surrénalectomie par un chirurgien expert, qui devra souvent réaliser un geste élargi aux organes de voisinage pour une exérèse tumorale en “monobloc”, seul moyen de minimiser le risque de récidive locorégionale. Ce risque persiste cependant, ainsi que le risque de métastases à distance : hépatiques, pulmonaires, osseuses, plus rarement cérébrales. Les lésions secondaires se développent à partir de micrométastases, déjà présentes lors du diagnostic et du traitement chirurgical : l’amélioration du pronostic des patients porteurs de CCS passe par un traitement systémique capable de prévenir leur développement. Pour le moment, un seul médicament a montré une certaine efficacité : le mitotane ou OP’DDD, dérivé de l’insecticide DDT, dont l’action toxique sur le cortex surrénalien a été identifiée dès l’après-guerre après diagnostic autopsique de chiens décédés d’insuffisance surrénale aiguë lors de campagnes de désinsectisation. L’effet inhibiteur de la stéroïdogénèse du mitotane est bien démontré (2), de même que son effet cytotoxique sur les cellules corticosurrénaliennes normales, même si celui-ci n’est pas forcément irréversible. Le mitotane est ainsi utilisé depuis longtemps pour contrôler les hypersécrétions des CCS, mais la question de son efficacité antitumorale reste encore controversée, qu’il s’agisse d’une action curative sur des métastases déjà présentes et visibles, ou “préventive” du développement de métastases chez des patients ayant bénéficié d’une chirurgie avec résection totale R0, sans métastases visibles, et chez lesquels le mitotane peut être donné en traitement adjuvant. En 2007, un travail multicentrique italo-allemand a donné des arguments forts en faveur de l’efficacité du mitotane en traitement adjuvant (1) : un groupe de patients italiens ayant bénéficié d’un traitement par mitotane adjuvant présentait un meilleur pronostic qu’un groupe de patients allemands et qu’un autre groupe de patients italiens, qui n’avaient pas été traités par mitotane. Le défaut de cette étude est qu’elle était rétrospective, ce qui expose à des biais qui limitent la portée des conclusions, qui restent discutées (3). Il faut cependant noter que, dans cette étude italo-allemande, la décision de traiter par mitotane était surtout dépendante d’un “effet centre” plutôt que de facteurs liés aux patients. Les facteurs pronostics des patients qui ont reçu le mitotane étaient d’ailleurs plus péjoratifs que ceux du groupe sans mitotane, biais qui ne devrait pas confondre les résultats positifs de l’étude. Une étude de suivi de ces patients vient d’être publiée et confirme ces résultats en faveur de l’efficacité du mitotane (4). Il faut admettre qu’une autre étude très récente, rétrospective multicentrique américaine, va dans le sens inverse (5) : on notera cependant que, dans cette dernière étude, les patients traités par mitotane avaient des facteurs pronostiques moins bons que les autres, notamment une fréquence de stade IV plus élevée, ce qui induit un biais difficile à corriger. Malgré ces dissonances persistantes, un groupe d’experts internationaux reconnus a publié un consensus sur l’intérêt de traiter par mitotane les patients à haut risque de récidive (6), avec l’objectif d’obtenir des taux de mitotanémie > 14 mg/l (7). Le risque de récidive est évalué par la stadification proposée par le groupe européen ENSAT (8) [figure 1], et par le taux de prolifération tumorale mesurée par le Ki67 : pourcentage de cellules exprimant ce marqueur de prolifération, avec un seuil de 10 % au-delà duquel la tumeur est considérée comme à haut risque de récidive. Cette classification du risque de récidive est très bien décrite dans le thésaurus du cancer corticosurrénalien rédigé par le groupe français COMETE/Cancers de la surrénale, disponible gratuitement et sans inscription sur le site de la SFE (SFendocrino.org) par le chemin suivant : groupes-réseaux/COMETE-cancers de la surrénale/recommandations. La classification permet de définir 3 niveaux de risque de récidive : modéré, intermédiaire et fort. Figure 1. Stades TNM et ENSAT des cancers corticosurrénaliens
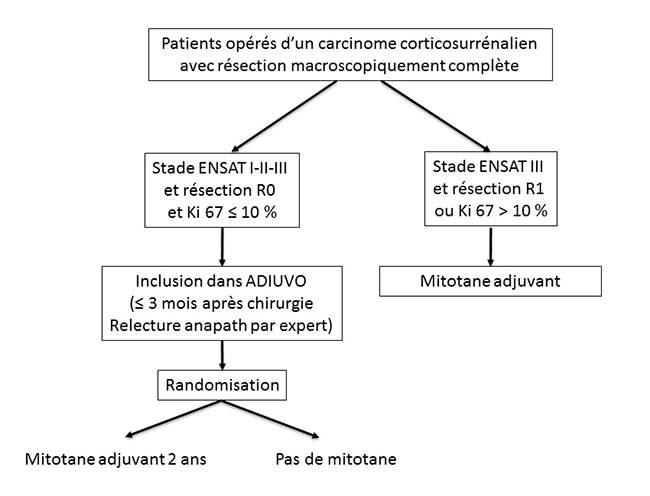
Le consensus d’experts de 2010 propose qu’un traitement par mitotane soit proposé de façon systématique chez les patients du groupe à risque de récidive fort, et sur une base “individuelle” (6) pour les patients des groupes de risque modéré et intermédiaire. C’est pour ces derniers patients que les groupes COMETE et ENSAT ont besoin de vous. En effet, c’est dans ces groupes qu’a enfin été lancée une étude prospective randomisée, type d’étude qui manque cruellement dans le domaine du CCS. C’est donc le grand mérite du Pr Massimo Terzolo, de l’université de Turin, d’avoir lancé en 2008 l’étude ADIUVO (https://www.epiclin.it/adiuvo), étude prospective randomisée multicentrique et internationale, qui propose aux patients opérés d’un CCS de risque de récidive modéré ou intermédiaire, avec Ki67 < 10 %, d’être inclus de façon randomisée soit dans un bras mitotane pendant 2 ans, soit dans un bras sans mitotane (figure 2). Figure 2. Étude ADIUVO : principaux critères d’inclusion
L’objectif principal d’ADIUVO est de déterminer si le mitotane prévient la survenue d’une récidive, le critère de jugement principal étant la survie sans progression. Il s’agit d’une étude universitaire où l’industrie pharmaceutique intervient pour fournir le mitotane et les dosages de mitotanémie aux patients. Le traitement par mitotane doit être commencé au plus tard 3 mois après la chirurgie. Cette étude européenne a pour coordonnateur français le Dr Éric Baudin, de l’institut Gustave-Roussy (baudin@igr.fr), et elle est également animée par la responsable du groupe COMETE-Cancers, de l’Institut national du cancer (INCa), le Dr Rossella Libe (rossella.libe@aphp.fr). Le groupe INCa COMETE-Cancers de la surrénale a besoin de vous ! Pour arriver à des résultats significatifs, l’étude ADIUVO a besoin de recruter 200 patients alors qu’aujourd’hui leur nombre reste insuffisant : 75 patients, dont 18 en France. Les raisons des difficultés de recrutement ont été analysées lors de la dernière réunion du groupe ADIUVO au congrès européen de Lisbonne (22 mai 2017) : bien sûr rareté du CCS, mais également problème d’un parti pris “pour” ou “contre” le mitotane par les médecins ou les patients, ces derniers pouvant de plus avoir du mal avec le concept d’être tirés au sort pour recevoir un traitement qui n’est pas dénué d’effets indésirables. Il n’en reste pas moins que ce protocole est le seul moyen de savoir si le traitement par mitotane offre un bénéfice en termes de survie sans récidive (et de survie tout court). Du fait de la rareté du CCS, beaucoup de centres qui participent à ADIUVO n’ont inclus qu’un tout petit nombre de patients, voire un seul ou pas du tout : si vous avez un patient concerné, pensez à ADIUVO et rapprochez-vous du responsable national de cette étude multicentrique ! Pour plus d’information : |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Références bibliographiques 1. Terzolo M, Angeli A, Fassnacht M et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356(23):2372-80. 2. Luton JP, Cerdas S, Billaud L et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med 1990;322(17):1195-201. 3. Huang H, Fojo T. Adjuvant mitotane for adrenocortical cancer–a recurring controversy. J Clin Endocrinol Metab 2008;93(10):3730-2. 4. Berruti A, Grisanti S, Pulzer A et al. Long-term outcomes of adjuvant mitotane therapy in patients with radically resected adrenocortical carcinoma. J Clin Endocrinol Metab 2017;102(4):1358-65. 5. Postlewait LM, Ethun CG, Tran TB et al. Outcomes of adjuvant mitotane after resection of adrenocortical carcinoma: a 13-institution study by the US Adrenocortical Carcinoma Group. J Am Coll Surg 2016;222(4):480-90. 6. Berruti A, Fassnacht M, Baudin E et al. Adjuvant therapy in patients with adrenocortical carcinoma: a position of an international panel. J Clin Oncol 2010;28(23):e401-2; author reply e3. 7. Hermsen IG, Fassnacht M, Terzolo M et al. Plasma concentrations of o,p’DDD, o,p’DDA, and o,p’DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab 2011;96(6):1844-51. 8. Fassnacht M, Johanssen S, Quinkler M et al. Limited prognostic value of the 2004 International Union against cancer staging classification for adrenocortical carcinoma: proposal for a revised TNM classification. Cancer 2009;115(2):243-50.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
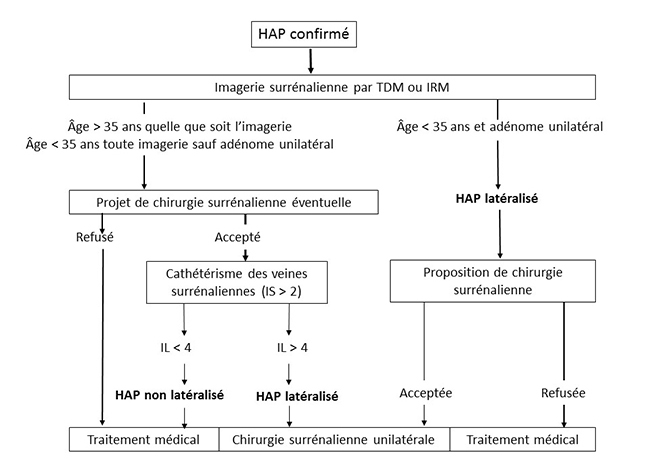
SPARTACUS bouscule les consensus Spartacus est le nom d’un esclave d’origine thrace qui se révolta contre le pouvoir romain en 73 avant Jésus-Christ, mais depuis peu c’est aussi le nom d’une étude hollando-polonaise qui fait trembler les consensus et les experts de l’hyperaldostéronisme primaire (HAP) : Subtyping Primary Aldosteronism: a Randomized Trial Comparing Adrenal Vein Sampling and Computed Tomography Scan, dont les résultats ont été publiés récemment (1). Comme son nom l’indique, SPARTACUS est une étude prospective randomisée, véritable révolution dans le domaine de l’HAP, qui vise à comparer les performances du cathétérisme des veines surrénaliennes (CVS) et du scanner surrénalien (tomodensitométrie [TDM]) pour le diagnostic étiologique de l’HAP. Nous sommes ici chez des patients dont l’HAP a déjà été démontré, pour lesquels il s’agit de déterminer s’il est latéralisé (comme dans le classique adénome de Conn), ce qui permet d’envisager un traitement chirurgical par surrénalectomie unilatérale, ou si il ne l’est pas (comme dans l’hyperplasie bilatérale), ce qui ne laisse que l’option d’un traitement médical. En effet, la surrénalectomie unilatérale est réputée ne pas pouvoir améliorer l’HAP si celui-ci est lié à une hyperplasie bilatérale avec sécrétion équivalente de chaque surrénale, même si en fait ce point peut être discuté (cf. ci-dessous [2]). Jusqu’ici, le dogme, repris dans les consensus américains (3, 4) et français (5) de l’HAP, était que le scanner surrénalien n’est pas fiable en raison de la fréquence des adénomes surrénaliens non sécrétants : un nodule surrénalien visible en TDM a beaucoup de chances de ne pas être un adénome de Conn, mais plutôt un banal adénome non sécrétant, chez un patient qui a par ailleurs un HAP lié à une hyperplasie bilatérale. Fonder une surrénalectomie sur la TDM expose donc à ne pas guérir les patients d’un HAP. Comme la fréquence des adénomes non sécrétants augmente avec l’âge, le risque d’erreur de la TDM augmente aussi avec celui-ci, et il faut que le patient soit jeune pour que ce risque devienne négligeable : les différents consensus mettent la barre à 35 ans, âge en dessous duquel on peut considérer la TDM comme fiable si elle met en évidence une image d’adénome unilatéral. Au-dessus de 35 ans, ce qui est de loin le cas le plus fréquent, il faut donc toujours faire appel au CVS, exploration techniquement complexe, réservée à des radiologues très entraînés, qui permet d’évaluer la contribution de chaque surrénale à l’hypersécrétion d’aldostérone. Ces considérations sur les valeurs diagnostiques de la TDM et du CVS et sur l’effet de la surrénalectomie unilatérale sont le fondement des algorithmes défendus dans les consensus actuels, y compris l’excellent consensus français de la SFE/SFHTA/AFCE, en accès libre sur PubMed (5) [figure 1] ou disponible en version française sur le site de la SFE (SFendocrino.org). Figure 1. Consensus français sur l’HAP
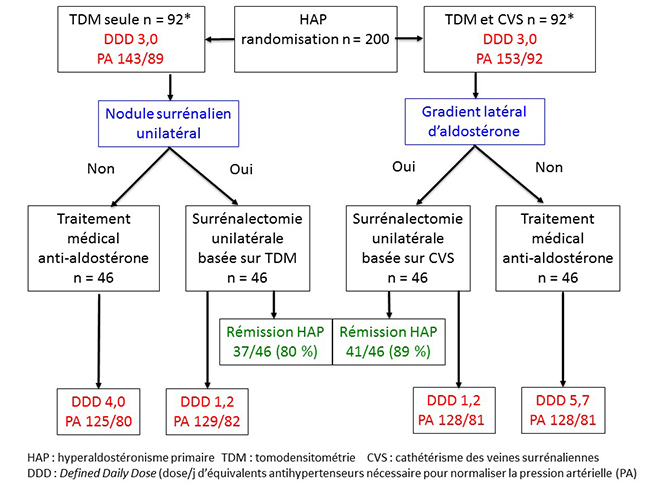
L’interêt de SPARTACUS est non seulement son caractère prospectif mais surtout la façon dont les auteurs résolvent le problème de l’absence de gold standard diagnostique pour comparer CVS et TDM : de façon pragmatique, ils décident de les comparer sur les résultats de la surrénalectomie unilatérale, décidée soit sur le CVS soit sur la TDM. Ces résultats sont analysés de 2 façons : le bénéfice tensionnel et la rémission de l’hyperaldostéronisme primaire. Pour analyser le bénéfice tensionnel, les auteurs utilisent une technique d’évaluation devenue courante dans le domaine de l’HTA, qui est la mesure du DDD (Daily Dose Delivery) : il s’agit simplement de compter les équivalents de traitements antihypertensifs nécessaires pour normaliser la tension artérielle d’un patient. Une intervention thérapeutique qui a un bénéfice tensionnel se traduira forcément par une baisse de la DDD. Pour calculer celle-ci, des équivalents ont été établis entre les différents traitements antihypertensifs, prenant en compte non seulement le type d’hypertenseur mais aussi sa dose. Bien évidemment, on ne peut comparer les DDD que chez des patients de même niveau tensionnel, mais dans l’étude SPARTACUS les auteurs ont parfaitement réussi à contrôler la tension artérielle aux mêmes niveaux dans les groupes CVS et TDM avant et après la surrénalectomie unilatérale (figure 2). Figure 2. Étude Spartacus : rémission de l’HAP et bénéfice tensionnel
La surprise de SPARTACUS est que le bénéfice tensionnel est le même, que la surrénalectomie unilatérale soit décidée sur la TDM ou sur le CVS (figure 2) ! Ce bénéfice est d’ailleurs tout à fait intéressant, avec une baisse moyenne de 1,8 de la DDD pour des résultats tensionnels meilleurs en postopératoire qu’en pré-opératoire. L’autre façon d’évaluer le bénéfice de la surrénalectomie est la détermination de la fréquence de rémission de l’hyperaldostéronisme primaire : en se basant sur des explorations classiques (mesure d’aldostérone et de rénine à l’état basal et après freinage par charge sodée intraveineuse), les auteurs retrouvent une rémission de l’HAP de 80 % dans le bras TDM contre 89 % dans le bras CVS, différence non significative (figure 2). Au total, dans cette étude, les résultats de la surrénalectomie unilatérale sont tout à fait intéressants, mais surtout ils sont équivalents, que le choix de la surrénale à réséquer se fasse sur la TDM ou sur le CVS. Ces résultats inattendus ont bien sûr bousculé les experts qui ne faisaient pas partie de cette étude hollando-polonaise : comment expliquer que le choix basé sur la TDM fasse aussi bien que le choix basé sur le CVS alors que, selon le dogme, la TDM est destinée à se fourvoyer en raison des adénomes non sécrétants ? Le débat agite actuellement la communauté de l’HAP, et des critiques tout à fait justifiées ont été avancées ; elles portent notamment sur la puissance de l’étude, qui reste limitée, et sur la technique utilisée pour le CVS (cathétérisation successive sous stimulation par ACTH) alors que d’autres centres prônent le cathétérisme bilatéral simultané sans ACTH (6). Une session entière est consacrée aux conclusions à donner à cette étude au congrès international sur l’HAP qui se tient prochainement à Munich (PIPA 5, 3-4 juillet 2017). Nous verrons bien quelles en seront les conclusions. Cependant, on peut naïvement relever les points suivants : SPARTACUS peut faire penser que la TDM ferait aussi bien que le CVS. Or il semble établi que la présence d’adénomes non sécrétants permet dans certains cas au CVS d’être plus performant que la TDM (cf. supra) : si malgré cela TDM et CVS sont en fait équivalents, alors il doit nécessairement y avoir des patients chez lesquels le CVS fait moins bien que la TDM, mais comment est-ce possible ? (Nous pensons avons trouvé une explication, qui sera développée ultérieurement.) Enfin, l’autre question naïve est la suivante : est-on vraiment sûr que la surrénalectomie unilatérale ne “marche pas” dans les hyperplasies bilatérales lorsque la sécrétion de chaque surrénale est équivalente ? En effet, si elle “marchait”, il serait facile d’expliquer que, malgré les adénomes non sécrétants, les résultats de la surrénalectomie unilatérale basée sur la TDM puissent être aussi bons que ceux de la surrénalectomie unilatérale basée sur le CVS. Il n’y a que très peu d’études où les auteurs ont osé faire des surrénalectomies unilatérales chez des patients qui ne montraient pas de latéralisation mais, de façon remarquable, une de ces études donne des résultats qui confirment une efficacité intéressante (2) ! Pour finir, une consolation pour les aficionados du CVS (dont nous faisons partie !) : lorsqu’il n’y a pas de nodule surrénalien détectable en TDM, le CVS reste le seul moyen de décider du côté de la surrénalectomie. Les équipes qui pratiquent de façon systématique le CVS savent bien que des patients porteurs d’HAP avec des surrénales d’aspect normal peuvent montrer une latéralisation très nette de la sécrétion d’aldostérone. Il semble quand même logique d’enlever alors la surrénale qui sécrète le plus d’aldostérone : pour ces patients, le CVS va continuer à être intéressant même si l’on finit par conclure à la validité de SPARTACUS pour les autres. À suivre ! |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Références bibliographiques 1. Dekkers T, Prejbisz A, Kool LJ et al. Adrenal vein sampling versus CT scan to determine treatment in primary aldosteronism: an outcome-based randomised diagnostic trial. Lancet Diabetes Endocrinol 2016;4(9):739-46. 2. Sukor N, Gordon RD, Ku YK et al. Role of unilateral adrenalectomy in bilateral primary aldosteronism: a 22-year single center experience. J Clin Endocrinol Metab 2009;94(7):2437-45. 3. Funder JW, Carey RM, Fardella C et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008;93(9):3266-81. 4. Funder JW, Carey RM, Mantero F et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016;101(5):1889-916. 5. Amar L, Baguet JP, Bardet S et al. SFE/SFHTA/AFCE primary aldosteronism consensus: introduction and handbook. Ann Endocrinol (Paris) 2016;77(3):179-86. 6. Rossi GP, Funder JW. Adrenal venous sampling versus computed tomographic scan to determine treatment in primary aldosteronism (The SPARTACUS trial): a critique. Hypertension 2017;69(3):396-7.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Génétique des phéochromocytomes et des paragangliomes pour les nuls : Qu’il est loin le temps où les phéochromocytomes observaient la règle des 10, qui comprenait notamment 10 % de formes familiales ! Il s’agissait alors de 3 gènes, RET, VHL et NF1, correspondant à des maladies rares mais assez bien connues. Depuis, la liste des gènes prédisposant aux phéochromocytomes et/ou paragangliomes (PPGL) s’est bien allongée, et il est bien difficile pour le clinicien de les connaître tous : des mutations germinales de… 17 gènes ont été décrites chez des patients porteurs de phéochromocytomes, éventuellement associés à d’autres lésions (auxquelles il faut rajouter 2 gènes présentant des mutations post-zygotiques en mosaïque). Mutations germinales décrites dans les phéochromocytomes et les paragangliomes
Listes des gènes porteurs de mutations somatiques dans les PPGL :
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Références bibliographiques 1. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015;11(2):101-11. 2. NGS in PPGL Study Group, Toledo RA, Burnichon N, Cascon A et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol 2017;13(4):233-47. 3. Lussey-Lepoutre C, Buffet A, Gimenez-Roqueplo AP et al. Mitochondrial deficiencies in the predisposition to paraganglioma. Metabolites 2017;7(2).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Action non génomique des glucocorticoïdes dans les cellules hypophysaires gonadotropes Commentaires sur l’article “A novel non genomic glucocorticoid signaling mediated by a membrane palmitoylated glucocorticoid receptor cross talks with GnRH in gonadotrope cells”, de M. Ayrout, V. Simon, V. Bernard et al. (unité INSERM 1185, Le Kremlin-Bicêtre), accepté pour publication dans la revue Scientific Reports du groupe Nature en 2017. Nous avons tous appris que, comme les autres stéroïdes, le cortisol se lie à un récepteur, le récepteur glucorticoïde (GR), qui a des actions génomiques au niveau du noyau. En effet, le GR est une protéïne présente dans le cytoplasme qui, lorsqu’elle lie le cortisol, peut migrer dans le noyau et se lier à des séquences d’ADN pour modifier l’expression de certains gènes. Le GR se comporte ainsi comme un facteur de transcription activé par sa liaison à l’hormone, mécanisme qui rend compte de la plupart des actions du cortisol. Mais il existe des exceptions à cette règle : depuis quelques années ont été mises en évidence des actions du cortisol qui ne semblent pas s’expliquer par ce mécanisme, car elles sont trop rapides pour dépendre de la variation de l’expression des gènes et persistent même si l’on inhibe la transcription. Le mécanisme de ces actions, appelées “non génomiques”, reste controversé, mais l’hypothèse favorisée est qu’elle impliquerait un GR membranaire, issu du même gène que le classique GR nucléaire. L’intérêt de l’article que nous présentons ici est de mettre en évidence puis d’analyser en détail une de ces actions non génomiques des glucocorticoïdes, l’inhibition de la sécrétion de la cellule hypophysaire gonadotrope. Nous savons qu’une hypersécrétion de cortisol inhibe la sécrétion de gonadotrophines, car ceci rend compte de l’insuffisance gonadotrope fonctionnelle liée au stress. Jusqu’ici, le mécanisme généralement invoqué pour cette action du cortisol est une action inhibitrice classique sur les neurones à GnRH de l’hypothalamus. Cependant, il a été mis en évidence in vivo, dans un modèle animal, une action rapide du cortisol d’inhibition des pics de LH, induit par la sécrétion pulsatile de GnRH, ce qui suggère une action hypophysaire et rapide du cortisol. Dans cet article, M. Ayrout et al. donnent plusieurs arguments pour l’implication effective d’une action non génomique des glucocorticoïdes sur les cellules hypophysaires gonadotropes. Leur modèle expérimental est une lignée cellulaire hypophysaire gonadotrope LbT2 d’origine murine (on rappelle que chez la souris le glucocorticoïde physiologique n’est pas le cortisol mais la corticostérone), dans laquelle la stimulation par le GnRH induit une sécrétion de LH. En utilisant ce modèle, les auteurs démontrent en effet que la corticostérone induit sur les cellules gonadotropes une stimulation très rapide, en moins de 5 minutes, de la phosphorylation de 2 protéines : la protéine kinase calmoduline-dépendante (CaMKII) et la synapsine 1. Or la CaMKII est précisément considérée comme une étape importante dans la stimulation de la sécrétion des gonadotrophines par le GnRH. Quels sont les mécanismes de cette action de la corticostérone ? M. Ayrout et al. donnent des arguments solides en faveur d’une activation par un GR localisé non pas dans le noyau mais dans la membrane plasmique. En effet, cette action est observée même avec de la corticostérone liée à une grosse protéine, la sérum albumine bovine, qui ne lui permet pas de traverser la membrane plasmique. Les auteurs démontrent qu’il y a dans les cellules LbT2 une expression du GR dans la membrane plasmique, et un point très intéressant est que ce GR semble lié à la membrane plasmique par l’intermédiaire d’une chaîne d’acides gras palmitiques. En effet, l’inhibition de la palmitylation aboutit à une perte de l’expression membranaire du GR et à une perte de l’effet de la corticostérone sur la phosphorylation de la CaMKII. Quels liens peut-on faire entre cette action hypophysaire non génomique des glucocorticoïdes et la sécrétion de gonadotrophines induites par le GnRH ? Pour répondre à cette question, les auteurs étudient l’effet des glucocorticoïdes sur la phosphorylation de la CaMKII par le GnRH : ils retrouvent que, malgré l’effet propre des glucocorticoïdes sur la CaMKII, ils inhibent très rapidement la stimulation de la CaMKII induite par le GnRH. Ceci offre un mécanisme possible de l’action inhibitrice rapide des glucocorticoïdes sur la sécrétion de gonadotrophine induite par le GnRH. Bien sûr, il faut enfin aussi faire un lien entre ces événements moléculaires précoces et la survenue, plus tardive, d’une sécrétion de LH, qu’il n’est pas possible d’observer dans les conditions expérimentales précédentes. Pour cela, les auteurs utilisent un autre modèle, des explants d’hypophyse murine stimulés in vitro par du GnRH pendant 1 heure, où ils montrent que l’ajout dans le milieu de culture de dexaméthasone entraîne une inhibition significative de la sécrétion de LH induite par le GnRH, alors qu’elle n’induit pas d’effet sur l’expression du gène de la LH. Au total, le travail de l’équipe du Kremlin-Bicêtre permet de conclure que l’effet inhibiteur des glucocorticoïdes sur la sécrétion de gonadotrophines implique un effet rapide non génomique, qui passe par l’activation d’un GR membranaire. L’intérêt de ce travail est qu’il élargit notre vision de l’action des glucocorticoïdes : cette action ne se limite pas à une modulation de la transcription de gènes cibles mais implique aussi les voies de signalisation induites par des récepteurs membranaires.
L’auteur déclare avoir des liens d’intérêts avec Ipsen, Novartis et Pfizer. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Vous recevez cette newsletter car vous vous êtes inscrit sur le site de la SFE. Si vous ne voulez plus recevoir cette newsletter, cliquez sur le lien suivant Désinscription. Conformément à la loi Informatique et Libertés du 06/01/1978, vous disposez d’un droit d’accès, de rectification et d’opposition aux informations vous concernant qui peut s’exercer par courrier à : SFE 88, rue de la Roquette – 75011 – Courriel : webmaster@sfendocrino.org |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||