
| Accédez au site |
Accédez au |
 |
Lettre N°5 - Juillet 2012 |
[ÉDITORIAL] Avec l'identification de MAX, le phéochromocytome devient la tumeur neuroendocrine à 10 gènes de prédisposition. Nouvelles recommandations [CAS CLINIQUES] |
• Méfions-nous des petites cellules ! [L'ACTUALITÉ COMMENTÉE]
|
Avec l'identification de MAX, le phéochromocytome devient la tumeur neuroendocrine à 10 gènes de prédisposition. Nouvelles recommandations
Anne-Paule Gimenez-Roqueplo (Paris)
Après TMEM127, identifié en 2010 (1), qui contribue à environ 1,6 % des formes héréditaires de la maladie (2), MAX est le dixième gène de prédisposition au phéochromocytome. Il a été découvert en 2011 par l’équipe de Mercedes Robledo (Centro Nacional de Investigaciones Oncológicas [CNIO], Madrid) grâce à une stratégie de séquençage d’exomes menée chez 3 patients atteints ayant des antécédents familiaux de phéochromocytome (3). Très rapidement après cette découverte, un effort international sans précédent, mobilisant un grand nombre de centres en France (GTE, INCa-COMETE et RENATEN) et à travers le monde (fédérés sous l’égide du réseau européen ENS@T [European Network for the Study of Adrenal Tumors] et du Familial Pheochromocytoma Consortium aux États-Unis), a permis de rapidement établir l’intérêt clinique de ce nouveau gène (4). Parmi les 1694 patients inclus, qui ont tous eu au préalable un génotypage négatif des principaux gènes de prédisposition, 19 patients (1,12 %) se sont avérés porteurs d’une mutation constitutionnelle de MAX, dont seuls 7 (37 %) avaient des antécédents familiaux connus de phéochromocytome. Dans tous les cas, la maladie avait été révélée par une tumeur développée aux dépens de la glande surrénale, unique, bilatérale ou multiple sur la même glande. L’âge moyen au diagnostic était de 34 ans. Trois patients ont développé ensuite un paragangliome, et 2, des métastases. Alors que la contribution des mutations somatiques de RET et de VHL dans la tumorigenèse du phéochromocytome a récemment été décrite comme significative (15 %) [5], une mutation somatique de MAX a également été retrouvée dans 5 tumeurs sur 245 analysées (1,65 %).
D’une tumeur à 10 % de formes génétiques, le phéochromocytome est passé, en une dizaine d’années, à une tumeur à 10 gènes de prédisposition dont les mutations, constitutionnelles ou somatiques, expliquent près de la moitié des cas. Pour les laboratoires qui réalisent en routine le test génétique du phéochromocytome, le profil de sécrétion catécholaminergique de la tumeur et les résultats des analyses immunohistochimiques ciblant SDHB et MAX s’avèrent être désormais des éléments importants pour guider l’analyse génétique. Ainsi, cette étude a montré, d’une part, que les tumeurs dont le gène MAX est inactivé, bien que capables de produire de l’adrénaline, sécrètent de préférence de la noradrénaline et, d’autre part, que l’immunohistochimie MAX permet de détecter les tumeurs ayant une mutation entraînant la production d’une protéine MAX tronquée. L’ensemble de ces nouvelles données a été pris en compte par le réseau des laboratoires d’oncogénétique des tumeurs endocrines rares pour établir l’algorithme décisionnel des explorations génétiques des phéochromocytomes et des paragangliomes qui est disponible sur le site Web du GTE (6).
Références bibliographiques
1. Qin Y, Yao L, King EE et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 2010;423:229-33.
2. Abermil N, Guillaud-Bataille M, Burnichon N et al. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J Clin Endocrinol Metab 2012;97:E805-9.
3. Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011;43:663-7.
4. Burnichon N, Cascón A, Schiavi F et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 2012;18:2828-37.
5. Burnichon N, Vescovo L, Amar L et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 2011;20:3974-85.
6. http://www.sfendocrino.org/article/249/recommandations

De l'intérêt de la TEP à la F-dopa en cas de suspicion clinique et biologique de phéochromocytome sans image visible en imagerie conventionnelle
Juliette du Payrat (Lyon), Françoise Borson-Chazot (Lyon)
L’histoire de Mme B. débute en 1955, avec la découverte d’une hypertension artérielle à l’âge de 19 ans, sans histoire familiale, ni antécédents personnels particuliers. Le bilan met en évidence une élévation des dérivés méthoxylés conduisant au diagnostic d’un phéochromocytome, traité par surrénalectomie gauche. Les dérivés méthoxylés se normalisent après l’opération. Trente-deux ans plus tard, en 1987, on constate une récidive clinique et biologique amenant à la découverte d’une lésion surrénalienne droite. Celle-ci est alors traitée par une surrénalectomie droite, qui met en évidence des lésions surrénaliennes multifocales. Le bilan postopératoire confirme la guérison avec normalisation des dérivés méthoxylés.
À partir de 2003, la normétadrénaline s’élève progressivement, atteignant 7 859 ng/l (1 000 < N < 2 000), la métadrénaline restant normale.
Le bilan morphologique, qui comprend alors un scanner thoracoabdominal, une scintigraphie à la MIBG, un Octreoscan®, et une TEP au FDG, reste négatif.
Il est finalement complété par une TEP à la F-dopa qui met en évidence une masse hypermétabolique hypogastrique (figure), retrouvée sur un scanner orienté sous la forme d’une masse interaorticocave. Son exérèse confirme qu’il s’agit d’un paragangliome de 20 mm, appendu à un ganglion sympathique.
Le bilan postopératoire montre la normalisation de la normétadrénaline à 1 750 ng/l.
Une recherche génétique est réalisée, qui ne retrouve pas de mutation des gènes RET et SDH, mais une mutation ponctuelle faux-sens de l’exon 3 du gène VHL, caractéristique des maladies de Von Hippel-Lindau de type 2, ou formes avec phéochromocytome.
Cette observation illustre l’étude récente de Weisbrod et al. (1) rapportant une série de 52 patients suivis de façon prospective pour une maladie de Von Hippel-Lindau à Bethesda. La sensibilité de la TEP à la F-dopa y est comparée à celle de l’imagerie conventionnelle (TDM et IRM), et de la TEP au FDG. La TEP à la F-dopa ne visualise que 40 des 139 lésions mises en évidence. Néanmoins, 9 de ces lésions ne sont visibles que sur cet examen (4 lésions surrénaliennes, 2 lésions hépatiques et 3 lésions pancréatiques). C’est par ailleurs la technique d’imagerie qui a mis en évidence le plus de lésions surrénaliennes dans cette série.
En conclusion, la TEP à la F-dopa ne peut se substituer aux autres modalités d’imagerie dans la maladie de Von Hippel-Lindau, mais trouve son utilité en cas de suspicion de lésion surrénalienne ou de localisations atypiques.
Référence bibliographique
1. Weisbrod AB, Kitano M, Gesuwan K et al. Clinical utility of functional imaging with 18F-FDOPA in Von Hippel-Lindau syndrome. J Clin Endocrinol Metab 2012;97:E613-7.
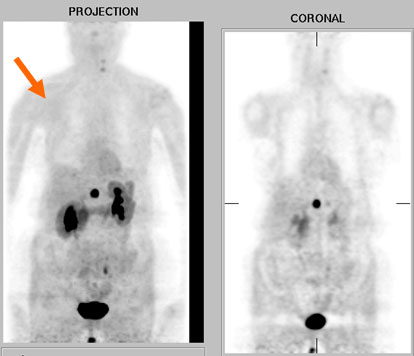
Figure. TEP à la F-dopa : mise en évidence d'une masse hypermétabolique hypogastrique.
Méfions-nous des petites cellules !
Cécile Chougnet (Villejuif), Éric Baudin (Villejuif)
Une masse cérébrale est découverte après une chute avec perte de connaissance chez un patient de 61 ans sans antécédent notable, à part un éthylisme chronique. Les premiers examens retrouvent une fracture du plancher de l’orbite et une tumeur de 22 × 25 mm de la région sphénoïdale centrée sur le sinus sphénoïdal et la selle turcique, avec anomalie du signal du corps sphénoïdal, envahissant le sinus caverneux gauche sans destruction osseuse. Le patient est transféré dans un service de neurochirurgie pour bénéficier d’une biopsie transsphénoïdale. L’examen histologique montre une tumeur à petites cellules rondes et basophiles largement remaniée par de la nécrose tumorale. Les atypies et l’anisonucléose sont, dans l’ensemble, discrètes à modérées. Il n’est pas observé d’images de mitoses (dans la limite du prélèvement très peu abondant). Les immunomarquages montrent que les cellules sont cytokératines AE1/AE3 ++ d’aspect granuleux, CK7 positif dans 2-3 cellules, CD56 ++, chromogranine ++ ; synaptophysine ++, NSE ++, CD99 +, EMA −, TTF1 −, PS100 −. Le diagnostic de carcinome endocrine à petites cellules est posé, et confirmé dans un deuxième centre.
La TEP au FDG montre un hypermétabolisme modéré au niveau de la masse sphénoïdale (SUV 5,4) et d’une masse paracardiaque droite suspecte (SUV 5,1). La fibroscopie bronchique ne retrouve pas de lésion endobronchique visible.
Le patient est donc adressé pour une chimiothérapie de type platine VP16 du fait de ce diagnostic de carcinome endocrine à petites cellules, possiblement de primitif bronchique avec métastase hypophysaire.
Cliniquement, le champ visuel est normal, mais le patient présente une désorientation temporospatiale, une tendance à la persévération, un ralentissement psychomoteur et des troubles de la mémoire. Le patient est en bon état général (OMS 1), les insuffisances corticotropes et thyréotropes sont correctement substituées, la prolactine est indétectable. Il ne présente pas de diabète insipide.
Devant la discordance entre l’absence de mitose et le caractère peu différencié, l’absence de destruction osseuse, la présence au contraire d’anomalies des fonctions anté- et posthypophysaire et la faible évolutivité en 3 mois de la lésion sphénoïdale, le diagnostic de métastase est remis en question. Une troisième relecture des lames de la biopsie est demandée. Celle-ci évoque un adénome hypophysaire. Le Ki67 est confirmé à 1 %.
La masse hypophysaire est retirée par exérèse transsphénoïdale. L’histologie révèle un adénome hypophysaire non sécrétant, exprimant fortement la chromogranine, faiblement la LH et la FSH ; les marqueurs TTF1, CK7, CK20, GH, prolactine, HCG, TSH et sous-unité α ne sont pas exprimés. Pas de mitose vue. Le Ki 67 est à 1 %.
Les carcinomes endocrines bronchiques peu différenciés peuvent donner des métastases hypophysaires (1). Mais il faut toujours se méfier du diagnostic de carcinome endocrine peu différencié, donné par excès, surtout lorsque le prélèvement est de petite taille ou de mauvaise qualité. En effet, les cellules des tumeurs neuroendocrines bien différenciées sur un petit prélèvement peuvent évoquer un carcinome à petites cellules (CPC) [2]. De plus, les données de la littérature montrent une reproductibilité entre anatomopathologistes du diagnostic de CPC de l’ordre de 95 %, tandis que le diagnostic de carcinome endocrine bronchique à grandes cellules est peu reproductible (3, 4). Les redressements diagnostiques vers les diagnostics finaux de tumeur neuroendocrine bien différenciée ne sont pas rares. La relecture de tous les carcinomes neuroendocrines peu différenciés doit être faite par un pathologiste du réseau TENpath en urgence.
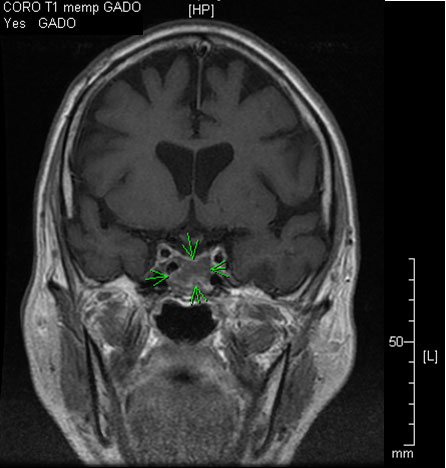
Figure. IRM : coupe coronale, T1, gadolinium.
Références bibliographiques
1. Faggiano A, Sabourin JC, Ducreux M et al. Pulmonary and extrapulmonary poorly differentiated large cell neuroendocrine carcinomas: diagnostic and prognostic features. Cancer 2007;110:265-74.
2. Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol 2010;21 Suppl 7:vii65-71.
3. Ha SY, Han J, Kim WS et al. Interobserver variability in diagnosing high-grade neuroendocrine carcinoma of the lung and comparing it with the morphometric analysis. Korean Journal of Pathology 2012;46:42-47.
4. Travis WD, Gal AA, Colby TV et al. Reproducibility of neuroendocrine lung tumor classification. Hum Pathol 1998;29:272-9.

IRM ou échoendoscopie pour l'évaluation initiale des tumeurs pancréatiques des NEM1 : les 2 sont complémentaires
Régis Cohen (Bobigny), Guillaume Cadiot (Reims)
Barbe C, Murat A, Dupas B et al.; Groupe d'étude des tumeurs endocrines (GTE). Magnetic resonance imaging versus endoscopic ultrasonography for the detection of pancreatic tumours in multiple endocrine neoplasia type 1. Dig Liver Dis 2012;44:228-34.
Pour les patients atteints de néoplasie endocrinienne multiple de type 1 (NEM1), le principal facteur de risque de métastases des tumeurs pancréatiques est leur taille. Les recommandations actuelles indiquent la chirurgie pour les tumeurs pancréatiques non fonctionnelles de plus de 20 mm. Le programme de surveillance pour les plus petites tumeurs est un premier contrôle à 1 an, pour éliminer tout risque de progression rapide, puis tous les 5 ans en l’absence de tumeur pancréatique identifiée, tous les 3 ans lorsque la ou les tumeurs mesurent moins de 10 mm ou enfin tous les 1 à 2 ans dans toutes les autres situations. Parce que l’échoendoscopie est invasive et nécessite une anesthésie générale, ce travail du groupe d’étude des tumeurs endocrines (GTE) a comparé les performances de l’échoendoscopie à celles de l’IRM pour les tumeurs de plus de 10 mm, avec pour objectif principal la concordance entre les 2 méthodes.
Cette étude prospective multicentrique a inclus 90 patients atteints de NEM1 avec lecture des images en aveugle et centralisée par des gastroentérologues et radiologues.
L’échoendoscopie a détecté plus de tumeurs que l’IRM (268 contre 158) ; sa supériorité a principalement concerné des lésions de petite taille. Elle a détecté 86 tumeurs de plus de 10 mm, chez 53,3 % des patients, contre 67 chez 51,1 % des patients pour l’IRM. Les résultats sont discordants. L’échoendoscopie et l’IRM ont visualisé 24 tumeurs de plus de 20 mm, mais elles en ont manqué respectivement 11/24 et 4/24. Elles ont détecté des tumeurs de plus de 10 mm chez 57 patients, mais ont été négatives pour les tumeurs de plus de 10 mm chez respectivement 9 et 11 patients. La localisation dans la queue du pancréas et le caractère isoéchogène (pauvre en graisse) étaient des causes fréquentes d’échec pour l’échoendoscopie. Le fait que l’échoendoscopie varie selon l’opérateur ou la prise de contraste insuffisante par gadolinium sont d’autres causes potentielles mais non totalement évaluables.
Il ressort que l’échoendoscopie et l’IRM sont donc complémentaires et doivent être effectuées toutes les deux à l’évaluation initiale des NEM1.
Attention : les données présentées ici, issues de la recherche, sont susceptibles de ne pas être validées par les autorités de santé françaises. Ces informations sont sous la seule responsabilité des auteurs qui sont garants de l'objectivité de cette publication.
