
| Accédez au site |
Accédez au |

En attendant les nouvelles recommandations du GTE, des recommandations internationales actualisées pour les néoplasies endocriniennes multiples de type 1 ?
Régis Cohen (Bobigny)
À chaque recommandation, le niveau de preuve est indiqué. Comme dans toute pathologie rare, il s’agit souvent de consensus d’experts.
Elles seront très utiles pour le clinicien et montrent que la recherche dans ce domaine reste plus que jamais nécessaire.
En attendant les prochaines recommandations du GTE, il s’agit d’une bonne mise au point, intégrant des références récentes émanant souvent, d’ailleurs, du GTE.
Donc, bonne lecture, et bonne rentrée !

Une tumeur endocrine trompeuse
Abdallah Al-Salameh (Bobigny)
La patiente, âgée de 64 ans, a consulté son médecin traitant pour asthénie. Le bilan initial retrouve une hyperparathyroïdie primaire (calcium : 2,78 mM, phosphore : 0,85 mM, parathormone : 232 pg/ml) et un goitre multinodulaire avec hypothyroïdie auto-immune.
Elle bénéficie d’une thyroïdectomie totale avec curage récurrentiel bilatéral (examen extemporané en faveur d’une lésion maligne) et d’une exploration des 4 parathyroïdes. On ne retrouve pas d’adénome mais le chirurgien enlève la parathyroïde inférieure droite. L’examen anatomopathologique initial conclut à une tumeur endocrine intrathyroïdienne bien différenciée de 1,5 cm (CgA+, CT-, TTF1- avec Ki-67 < 2 %), avec une parathyroïde normale.
En postopératoire survient une hypocalcémie profonde et prolongée (Ca : 1,63 mM, phosphore normal, PTH < 3 pg/ml, 25-OH-D : 24 ng/ml). Devant cette guérison sans adénome parathyroïdien, une relecture demandée confirme l’existence d’un adénome parathyroïdien intrathyroïdien (APIT), grâce à l’immunomarquage anti-PTH positif. Les tumeurs neuroendocrines de la thyroïde étant exceptionnelles, l’APIT est un diagnostic à connaître. En préopératoire, une fixation thyroïdienne à la scintigraphie MIBI doit faire discuter ce diagnostic, un dosage de PTH dans le liquide de rinçage de cytoponction peut le confirmer. En postopératoire, seul l’immunomarquage anti-PTH peut confirmer le diagnostic.
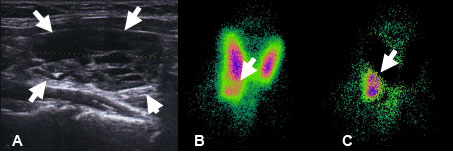
Figure. Échographie thyroïdienne (A) montrant le nodule de premier plan dans le pôle inférieur du lobe droit de la thyroïde. (B) et (C) Scintigraphie sestamibi couplée avec la scintigraphie thyroïdienne (iode) par soustraction montrant un nodule intrathyroïdien dans la partie inférieure du lobe droit.

Tumeurs neuroendocrines pancréatiques : intérêt d’un blocage simultané des voies de signalisation du VEGF et de MET pour prévenir les risques d’échappement aux traitements antiangiogéniques
Emmanuel Mitry (Paris)
Une des conséquences des traitements inhibiteurs de l’angiogenèse est la majoration de l’hypoxie intratumorale liée à la réduction vasculaire secondaire à l’inhibition du VEGF. Cette hypoxie intratumorale entraîne une majoration de l’expression tumorale de MET qui pourrait être associée à une majoration de l’agressivité tumorale et à une augmentation du risque de progression métastatique. La voie de signalisation HGF/MET favorise en effet la mobilité et la prolifération cellulaire, leur potentiel invasif et, donc, le risque de dissémination métastatique (1). L’activation de cette voie est un facteur de mauvais pronostic et de résistance au traitement, comme cela a été montré dans plusieurs types tumoraux. Les travaux publiés par l’équipe de D.M. McDonald avaient pour objectif d’estimer le risque de majoration de l’agressivité tumorale secondaire à l’utilisation de thérapies anti-VEGF, l’implication de la voie MET dans ce processus d’échappement et l’intérêt éventuel d’une inhibition sélective de MET dans la prévention de ces événements (2).
Le traitement antiangiogénique par anticorps anti-VEGF ou par sunitinib de modèles précliniques de tumeurs neuroendocrines pancréatiques (souris RIP-Tag2) entraîne une réduction du volume tumoral de 75 à 78 % avec, cependant, malgré cette diminution de taille, des signes d’agressivité locorégionale plus marqués et une augmentation du risque métastatique (figure 1). Les analyses réalisées suggèrent que cette plus grande agressivité tumorale était secondaire à une majoration de l’hypoxie tumorale liée à une moindre densité vasculaire (figure 2) induisant l’expression du facteur HIF-1α (Hypoxia-Inducible Factor 1 α) et de MET (3 fois plus élevée après traitement par anticorps anti-VEGF et 6 fois plus élevée après traitement par sunitinib) (figure 3).
Les tumeurs RIP-Tag 2 traitées par une molécule inhibitrice de MET (PF-04217903 ou PF-02341066 [crizotinib]), analysées après 14 à 17 semaines de croissance, présentaient approximativement la même taille que celles observées dans le groupe contrôle non traité. Cependant, leur agressivité locorégionale était moindre et le nombre et la taille des métastases hépatiques étaient également diminués. Dans ce même modèle tumoral, une inhibition simultanée de MET, par l’un des deux TKI anti-MET précédemment cités, et de VEGF, par un anticorps anti-VEGF ou le sunitinib, permettait d’améliorer le contrôle tumoral et majorait l’effet antiprolifératif et antimétastatique par rapport à un traitement par anti-MET seul. Des résultats comparables étaient observés dans un modèle d’adénocarcinome pancréatique (Panc-1) : l’expression de MET et l’agressivité des tumeurs Panc-1 étaient majorées par l’utilisation du sunitinib ; ces processus étaient bloqués par l’utilisation concomitante d’un inhibiteur de MET (PF-04217903).
Ces résultats suggèrent que le risque de majoration de l’agressivité tumorale après traitement antiangiogénique est dépendant de l’activité de MET, elle-même liée au degré d’hypoxie tumorale. Dans ce contexte, il y aurait un intérêt à bloquer simultanément les voies de signalisation de l’angiogenèse et celles de MET. C’est ce que fait une molécule comme le cabozantinib (XL184), une petite molécule inhibitrice de tyrosine kinase ciblant MET et VEGFR2, avec des premiers résultats encourageants dans des études précliniques (figure 4) et dans une étude de phase II de traitement des carcinomes médullaires de la thyroïde (3). Plusieurs études de phases II et III sont actuellement en cours avec cette molécule dans diverses localisations tumorales (carcinome médullaire de la tyroïde, prostate, ovaire, sein, carcinome hépatocellulaire).

Figure 1. Tumeurs RIP-Tag 2 : agressivité tumorale après traitement antiangiogénique (2).
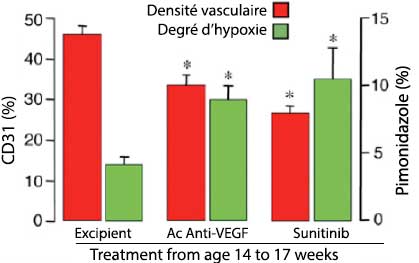
Figure 2. Tumeurs RIP-Tag 2 : densité vasculaire et degré d'hypoxie après traitement antiangiogénique (2).
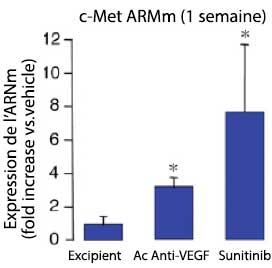
Figure 3. Tumeurs RIP-Tag 2 : expression de c-MET après traitement antiangiogénique (2).
![Figure 4. Tumeurs RIP-Tag2 : Risque invasif après traitement par cabozantinib (XL 184) [2]](http://www.sfendocrino.org/_newsletters/_images/gte/06-gte-fig4.jpg)
Figure 4. Tumeurs RIP-Tag 2 : risque invasif après traitement par cabozantinib (XL 184) (2).
Références bibliographiques
1. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003;4:915-25.
2. Sennino B, Ishiguro-Oonuma T, Wei Y et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov 2012;2:270-87.
3. Kurzrock R, Sherman SI, Ball DW et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 2011;29:2660-6.

Résumé des recommandations pour la prise en charge des patients atteints de
NEM 1
Abdallah Al-Salameh, Régis Cohen (Bobigny)
Thakker RV, Newey PJ, Walls GV et al. Clinical practice guidelines for Multiple Endocrine Neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990-3011.
| Description de la force des recommandations et de la qualité des preuves. Les recommandations fortes utilisent l'expression " nous recommandons " => numéro 1. Les recommandations faibles utilisent l'expression " nous suggérons " => numéro 2. De la même manière, les étoiles indiquent la qualité de la preuve, de sorte que : ∗ ∗∗ ∗∗∗ ∗∗∗∗ preuves de haute qualité. |
Résumé des recommandations
Recommandations générales
Les patients présentant une néoplasie endocrinienne multiple de type 1 (NEM1) et leurs familles doivent être pris en charge par une équipe multidisciplinaire composée de spécialistes compétents ayant une expérience dans le traitement des tumeurs endocrines (2 ∗∗![]()
![]() ).
).
Cette équipe devrait être composée de médecins spécialistes (par exemple, endocrinologue, gastroentérologue et oncologue) dans la gestion des tumeurs neuroendocrines (TNE), de chirurgiens endocriniens, d' anatomopathologistes (avec une expertise dans les TNE), de radiologues (y compris les médecins nucléaires) et de généticiens cliniques (2 ∗∗![]()
![]() ).
).
Tests génétiques
La recherche de mutations germinales de NEM1 doit être proposée aux cas index avec NEM1 et à leurs apparentés du premier degré (symptomatologique ou non) (1 ∗∗∗![]() ).
).
La recherche de mutations germinales de NEM1 des apparentés asymptomatiques doit être proposée dès l’âge de 5 ans (2 ∗∗![]()
![]() ).
).
La recherche de mutations germinales de NEM1 peut être recommandée chez les personnes ayant des phénotypes atypiques NEM1 (par exemple hyper-parathyroïdie avec hyperplasie) (2 ∗∗![]()
![]() ).
).
Tous les individus candidats à la recherche de mutations germinales de NEM1 doivent bénéficier d’une consultation génétique avant le test (1 ∗∗∗∗).
La recherche de mutations germinales de NEM1 doit être entreprise par un laboratoire labellisé en génétique clinique pour le gène NEM1 (1 ∗∗∗∗). Si la mutation NEM1 n’est pas identifiée dans une région codante, il faut rechercher des délétions partielles ou totales, analyser les haplotypes du locus NEM1, ou des gènes candidats connus (1 ∗∗∗![]()
![]() )
)
Dans la famille d’un patient atteint d’une NEM1 avec une mutation identifiée, l’analyse génétique doit toujours être réalisée avant les tests de dépistage biochimique et radiologique afin d’éviter des examens inutiles et coûteux (1 ∗∗∗![]() ).
).
Les personnes porteuses d’une mutation germinale NEM1 doivent être évaluées régulièrement (par exemple sur une base annuelle) pour le dépistage des tumeurs associées à la NEM1 (1 ∗∗∗![]() ).
).
Dépistage des tumeurs
Les personnes identifiées comme présentant un risque élevé de développement de tumeurs associées à la NEM1 (par exemple, cas index, porteurs d’une mutation NEM1) devraient bénéficier d’un programme de dépistage des tumeurs : clinique, biochimique et radiologique (détaillé ci-dessous). La nature et le moment du dépistage dépendent des ressources locales, du jugement clinique et des préférences du patient (2 ∗∗![]()
![]() ).
).
Tumeurs parathyroïdiennes
Diagnostic
Le dépistage de l’hyper-parathyroïdie primaire est réalisé annuellement par les dosages de la calcémie et de la PTH (1 ∗∗∗∗).
Traitement
La chirurgie effectuée par un chirurgien endocrinien expérimenté est le traitement de choix, même si le calendrier optimal n’a pas été défini. L’exploration conventionnelle bilatérale en chirurgie ouverte consiste en général en une parathyroïdectomie subtotale (au moins 3,5 glandes) ou une parathyroïdectomie totale (1 ∗∗∗![]() ). Parallèlement, la thymectomie transcervicale est également souhaitable au moment de la chirurgie (2 ∗∗
). Parallèlement, la thymectomie transcervicale est également souhaitable au moment de la chirurgie (2 ∗∗![]()
![]() ). La parathyroïdectomie totale avec autotransplantation peut être envisagée (2 ∗∗∗
). La parathyroïdectomie totale avec autotransplantation peut être envisagée (2 ∗∗∗![]() ). La parathyroïdectomie mini-invasive n’est généralement pas recommandée, car plusieurs glandes sont habituellement touchées (1 ∗∗∗
). La parathyroïdectomie mini-invasive n’est généralement pas recommandée, car plusieurs glandes sont habituellement touchées (1 ∗∗∗![]() ).
).
TNE pancréatique
Diagnostic
Le dépistage des TNE gastro-duodéno-pancréatiques doit inclure, au minimum, une évaluation annuelle à jeun des hormones gastro-intestinales, comprenant : la gastrine, le glucagon, le vaso-intestinal polypeptide, le polypeptide pancréatique, la chromogranine A, l’insuline avec une glycémie à jeun associée (2 ∗∗![]()
![]() ).
).
Un consensus de dépistage radiologique optimal n’a pas été établi et dépend des ressources locales, du jugement clinique et des préférences du patient. Un protocole minimal et annuel d’imagerie suggéré comprend une visualisation du pancréas et du duodénum par imagerie par résonance magnétique (IRM), ou par tomodensitométrie (TDM), ou par échoendoscopie (2 ∗∗![]()
![]() ).
).
Traitement
L’objectif principal est de maintenir les patients sans maladie et sans symptôme aussi longtemps que possible, avec une bonne qualité de vie (1 ∗∗∗∗).
L’objectif du traitement pour les personnes atteintes d’une TNE du pancréas symptomatique, comme l’insulinome, est de parvenir à la guérison, si possible par la chirurgie (1 ∗∗∗∗).
L’extension de la maladie devrait être évaluée de façon précise avant d’envisager un traitement spécifique (1 ∗∗∗![]() ).
).
Le traitement de référence du gastrinome reste controversé. La chirurgie d’un gastrinome pancréatique non métastasé peut être curative et doit être envisagée avec un chirurgien endocrinien expérimenté (2 ∗∗![]()
![]() ). Cependant, la plupart des patients atteints de NEM1 ont de multiples petits gastrinomes sous-muqueux duodénaux, et la prise en charge de ces tumeurs reste controversée. Nous suggérons une prise en charge médicale utilisant les inhibiteurs de la pompe à protons pour la plupart des patients (2 ∗∗
). Cependant, la plupart des patients atteints de NEM1 ont de multiples petits gastrinomes sous-muqueux duodénaux, et la prise en charge de ces tumeurs reste controversée. Nous suggérons une prise en charge médicale utilisant les inhibiteurs de la pompe à protons pour la plupart des patients (2 ∗∗![]()
![]() ). Cependant, dans les centres chirurgicaux expérimentés, l’exérèse locale de ces tumeurs avec curage ganglionnaire, duodénectomie, ou, moins fréquemment, une duodéno-pancréatectomie, peut également être discutée selon les préférences des patients, parce que de telles approches peuvent améliorer les taux de guérison (2 ∗∗
). Cependant, dans les centres chirurgicaux expérimentés, l’exérèse locale de ces tumeurs avec curage ganglionnaire, duodénectomie, ou, moins fréquemment, une duodéno-pancréatectomie, peut également être discutée selon les préférences des patients, parce que de telles approches peuvent améliorer les taux de guérison (2 ∗∗![]()
![]() ). Bien que la duodéno-pancréatectomie de Whipple offre les meilleures chances de guérison pour le gastrinome des NEM1, nous ne la suggérons pas pour la majorité des patients, car elle est associée à une mortalité opératoire et à une morbidité à long terme non négligeable, alors que les opérations moins invasives sont associées à une excellente survie à long terme (2 ∗∗∗
). Bien que la duodéno-pancréatectomie de Whipple offre les meilleures chances de guérison pour le gastrinome des NEM1, nous ne la suggérons pas pour la majorité des patients, car elle est associée à une mortalité opératoire et à une morbidité à long terme non négligeable, alors que les opérations moins invasives sont associées à une excellente survie à long terme (2 ∗∗∗![]() ).
).
Les approches médicales comprennent les inhibiteurs de la pompe à protons et les analogues de la somatostatine pour supprimer l’hyperacidité (1 ∗∗∗∗). Une surveillance périodique par fibroscopie est indiquée chez les personnes atteintes d’hypergastrinémie pour l’identification d’ulcères gastroduodénaux et la présence de carcinoïde gastrique (2 ∗∗![]()
![]() ).
).
Le rôle de la chirurgie du pancréas pour les tumeurs non fonctionnelles est controversé. Nous suggérons d’envisager la chirurgie pour des tumeurs qui sont d'une taille supérieure à 1 cm et/ou qui ont une croissance importante démontrée après 6 à 12 mois de surveillance (2 ∗∗![]()
![]() ).
).
Un anatomopathologiste spécialisé dans les TNE devrait examiner tous les tissus tumoraux. Les tumeurs doivent être classées en fonction de la classification OMS 2010 ; le stade doit être déterminé selon le score TNM de l’Union internationale contre le cancer (7e édition) et selon le stade T de la Société européenne des tumeurs neuro-endocrines en fonction du site tumoral (1 ∗∗∗∗).
Le traitement d’une masse tumorale non résécable comprend soit les analogues de la somatostatine, les biothérapies, la radiothérapie métabolique, les traitements locorégionaux et la chimiothérapie (1 ∗∗∗![]() ).
).
La chimiothérapie est indiquée pour les TNE pancréatiques inopérables ou métastatiques (1 ∗∗∗![]() ). Le sunitinib et l’évérolimus peuvent être envisagés chez les patients à un stade avancé (non opérable ou métastatique) pour une tumeur bien différenciée qui progresse (1 ∗∗∗
). Le sunitinib et l’évérolimus peuvent être envisagés chez les patients à un stade avancé (non opérable ou métastatique) pour une tumeur bien différenciée qui progresse (1 ∗∗∗![]() ).
).
Tumeurs hypophysaires
Diagnostic
Le dépistage hormonal des tumeurs hypophysaires dépend des ressources locales et du jugement clinique. Il peut comprendre une évaluation annuelle de la prolactine et de l’IGF-I (2 ∗∗∗![]() ), ainsi qu’une IRM hypophysaire tous les 3 à 5 ans (2 ∗∗
), ainsi qu’une IRM hypophysaire tous les 3 à 5 ans (2 ∗∗![]()
![]() ). Pour les patients ayant des anomalies hypothalamo-hypophysaires, d’autres dosages doivent être effectués pour mieux caractériser la nature des lésions hypophysaires et ses effets sur la sécrétion des autres hormones hypophysaires (1 ∗∗∗
). Pour les patients ayant des anomalies hypothalamo-hypophysaires, d’autres dosages doivent être effectués pour mieux caractériser la nature des lésions hypophysaires et ses effets sur la sécrétion des autres hormones hypophysaires (1 ∗∗∗![]() ).
).
Traitement
Le traitement des tumeurs hypophysaires associées aux NEM1 est similaire à celui des patients non-porteurs de NEM1 et se compose d’un traitement médical approprié (par exemple, agoniste de la dopamine pour le prolactinome ; octréotide ou lanréotide pour l’acromégalie) ou une hypophysectomie transsphénoïdale sélective, la radiothérapie étant réservée pour des tumeurs résiduelles non résécables (1 ∗∗∗∗).
TNE thymique, bronchopulmonaire et gastrique
Diagnostic
L’évaluation hormonale par les dosages de l’acide 5-hydroxyin-doleacetic urinaire et de la chromogranine A n’est pas utile (1 ∗∗∗∗).
Les scanners ou les IRM thoraciques tous les 1 à 2 ans sont recommandés pour la détection des tumeurs carcinoïdes bronchiques ou thymiques (2 ∗![]()
![]()
![]() ).
).
La fibroscopie gastrique (avec biopsies) tous les 3 ans chez les personnes atteintes d’hypergastrinémie pour la recherche d’ulcérations gastroduodénales ou de carcinoïdes de type II est recommandée (2 ∗∗![]()
![]() ). L’échoendoscopie et la scintigraphie aux analogues de la somatostatine peuvent aider au diagnostic (1 ∗∗∗∗).
). L’échoendoscopie et la scintigraphie aux analogues de la somatostatine peuvent aider au diagnostic (1 ∗∗∗∗).
Traitement
La chirurgie curative, lorsque cela est possible, est le traitement de choix pour les lésions thymiques et carcinoïdes bronchiques (1 ∗∗∗∗).
Lorsque la maladie est avancée et qu’une chirurgie curative n’est pas possible, d’autres thérapies comme la radiothérapie et la chimiothérapie peuvent être envisagées (2 ∗![]()
![]()
![]() ).
).
Le traitement optimal des carcinoïdes gastriques de type II n’a pas été établi. Pour les lésions de petite taille (< 10 mm), on peut envisager une surveillance endoscopique. Pour les tumeurs plus volumineuses, une résection endoscopique ou une gastrectomie partielle ou totale doit être envisagée. Les indications des analogues de la somatostatine dans le traitement des tumeurs carcinoïdes gastriques de type II ne sont pas définies (2 ∗∗![]()
![]() ).
).
Tumeurs de la surrénale
Diagnostic
Le dépistage doit comprendre au minimum un scanner abdominal ou une IRM tous les 3 ans (2 ∗∗![]()
![]() ). Les lésions surrénaliennes doivent rester sous surveillance radiologique et doivent être évaluées pour leur potentiel malin (1 ∗∗∗
). Les lésions surrénaliennes doivent rester sous surveillance radiologique et doivent être évaluées pour leur potentiel malin (1 ∗∗∗![]() ).
).
Les images doivent être examinées par un radiologue expérimenté dans l’imagerie des surrénales (1 ∗∗∗![]() ).
).
L’évaluation hormonale des lésions surrénaliennes doit être restreinte aux patients qui ont des manifestations cliniques ou des tumeurs de plus de 1 cm et devrait se concentrer sur l’évaluation pour l’hyperaldostéronisme primaire et l'hypercorticisme primaire (1 ∗∗∗![]() ).
).
Traitement
Le traitement des tumeurs surrénales associées à une NEM1 est semblable à celui des tumeurs surrénales non-NEM1. La chirurgie est indiquée pour les tumeurs fonctionnelles (par exemple, hyperaldostéronisme primaire ou hypercorticisme), et pour les tumeurs non fonctionnelles avec caractéristiques atypiques, de taille supérieure à 4 cm, ou ayant une croissance significative sur un intervalle de 6 mois (1 ∗∗∗![]() ).
).
Attention : les données présentées ici, issues de la recherche, sont susceptibles de ne pas être validées par les autorités de santé françaises. Ces informations sont sous la seule responsabilité des auteurs qui sont garants de l'objectivité de cette publication.
