La lettre Surrénales de Novembre 2015
|
Traitement prénatal par dexaméthasone dans l’HCS – HCS par déficit en 21-hydroxylase: contrôle hormonal, facteur déterminant de l’état de santé à l’âge adulte – Chirurgical du Cushing par hyperplasie surrénalienne macronodulaire bilatérale – MicroARNs et corticosurrénalome
|
||||||||
|
||||||||
|
Chers passionnés des surrénales : la Newsletter n° 6 ne peut pas vous laisser indifférents ! L’hyperplasie congénitale des surrénales (HCS) est à l’honneur, avec tout d’abord la brûlante controverse du traitement prénatal dans un article de synthèse, très équilibré, de l’équipe lyonnaise (V. Tardy-Guidollet) qui défend ce traitement, et que nous faisons précéder de la traduction du résumé de l’article à charge du Pr W. Miller, qui le pourfend. Le devenir à l’âge adulte d’une importante cohorte de filles et garçons porteurs d’HCS, est ensuite présenté par l’équipe de la Pitié-Salpétrière, dans un article qui reconnait les difficultés de la prise en charge mais qui est porteur d’espoir. L’hyperplasie macronodulaire bilatérale primitive a longtemps été au devant de la scène, pour la découverte de sa génétique, et avant cela pour ses fameux récepteurs ectopiques et leurs promesses thérapeutiques. Ici, c’est un traitement chirurgical beaucoup plus basique qui est défendu, avec des résultats étonnants qui permettent aux patients d’échapper à l’insuffisance surrénale définitive. L’insuffisance surrénale est justement le sujet d’un cas clinique présenté dans la rubrique « image » que nous faisons rimer avec « dosages ». Enfin le carcinome corticosurrénalien offre l’occasion de présenter l’intérêt clinique des microARN, petits ARN non codants qui ouvrent de nouvelles perspectives diagnostiques, et sans doute un jour thérapeutiques. Bonne lecture ! Nous demandons aux accros du phéo de nous pardonner, nous avons donné la primeur à la cortico, mais ce n’est que partie remise ! Enfin les lecteurs attentifs tiqueront peut-être sur le fort contenu grenoblois de cette lettre ? Une fois n’est pas coutume, nous demandons votre indulgence, nous voulions témoigner que nous ne sommes pas toujours sur les pistes… au moins en automne. |
||||||||
 |
||||||||
|
Le traitement prénatal par dexaméthasone dans l’hyperplasie congénitale des surrénales La thérapie fœtale de l’hyperplasie congénitale ne doit pas être faite (1) Résumé 1. Miller WL. Fetal endocrine therapy for congenital adrenal hyperplasia should not be done. Best Pract Res Clin Endocrinol Metab 2015;29(3):469-83. Bénéfices versus risques du traitement prénatal par dexaméthasone dans l’hyperplasie congénitale des surrénales : état des lieux actuel Un traitement prénatal par dexaméthasone (DEX) a été proposé il y a une trentaine d’années par l’équipe de pédiatrie lyonnaise aux couples à risque d’avoir un enfant atteint de forme classique d’hyperplasie congénitale des surrénales (risque de ¼ pour les parents d’un 1er enfant atteint de forme classique), pour prévenir la masculinisation des organes génitaux externes chez les filles atteintes (1). Son efficacité a été démontrée par des publications internationales, notamment françaises, suédoises et américaines, sur la réduction notoire voire la prévention de la virilisation des petites filles atteintes. L’efficacité implique un début avant 6 semaines de gestation (6 SG) ou 8 semaines d’aménorrhée (8 SA), une posologie de 20 ?g/kg/jour en 2 à 3 prises et un maintien du traitement jusqu’à la naissance en cas de fœtus féminin atteint. En l’absence de traitement prénatal, les petites filles atteintes bénéficient d’une chirurgie réparatrice et les quelques études publiées dans la littérature montrent des problèmes de sexualité à l’âge adulte chez les filles opérées il y a une vingtaine d’années. Cette chirurgie est à réserver à des chirurgiens extrêmement spécialisés pour obtenir des bons résultats et il faudra attendre quelques années pour évaluer les progrès des techniques chirurgicales plus récentes. Le problème posé par le traitement prénatal est qu’il n’est utile que pour les filles atteintes (1 fœtus sur 8). Afin de ne plus traiter inutilement les garçons, la détermination précoce du sexe fœtal sur sérum maternel ou test SRY a été validée en France en 2002 ; la prise en charge prénatale proposée en France (2) permet, grâce à la collaboration efficace entre cliniciens et biologistes, de ne plus traiter les garçons, tout en garantissant un traitement avant 6 SG chez les filles atteintes. Concernant les filles indemnes, l’exposition à la DEX est réduite grâce à un diagnostic prénatal précoce sur ponction de villosités choriales vers 10-11 SG, avec un rendu de résultat, en moins d’une semaine. Des questions ont été soulevées suite aux travaux réalisés chez les animaux et publiés depuis les années 1990. Une revue récente du Pr. W. Miller reprend des résultats obtenus à partir des expérimentations animales et des études épidémiologiques chez l’humain (3). Concernant les possibles risques de la DEX chez les enfants traités, nous estimons que les études sont rassurantes sur l’absence d’effets tératogènes, et que les malformations congénitales ne sont pas plus fréquentes chez les enfants HCS traités par DEX que dans la population générale. Le Pr Miller a rapporté un risque plus élevé de fentes palatines chez des enfants traités pendant la vie fœtale par des glucocorticoïdes (4). L’étude fondée sur des questionnaires remplis par des parents concernait l’ensemble des enfants avec une fente palatine répertoriés dans un registre national. On en déduit une élévation très modérée de fente palatine chez les enfants traités par glucocorticoïdes par rapport aux non traités, les glucocorticoïdes prescrits étant de nature différente de la DEX et utilisés à d’autres doses. Les auteurs concluaient à la nécessité d’études supplémentaires. À noter que ceci n’a pas été confirmé chez les enfants traités in utero par DEX en France, aux États-Unis et en Suède. Le Pr. W. Miller reprend également des résultats obtenus à partir des expérimentations animales avec une altération de l’hippocampe impliqué dans la mémoire et le comportement, troubles métaboliques (hypertension par atteinte rénale et hyperglycémie par atteinte pancréatique). À noter que le petit poids de naissance constaté chez les animaux serait en partie responsable de ces désordres métaboliques d’après les auteurs. Il est important de souligner que les doses de DEX utilisées chez l’animal sont 5 à 10 fois supérieures à celles utilisées chez l’humain et que dans nombre de ces études, la DEX est administrée à partir du milieu du 2e trimestre de la grossesse. Seules les filles atteintes sont traitées jusqu’à ce terme. Afin de répondre à la question de l’atteinte possible de l’hippocampe, la communauté médicale s’est penchée sur les aspects neurocognitifs des enfants traités in utero. L’équipe suédoise du Pr. S. Lajic a décrit en 2007 de possibles effets secondaires neurocognitifs seulement chez les enfants (garçons et filles) non atteints traités (5) : l’étude portait sur une petite cohorte d’enfants et concluait à l’absence de modification du QI et d’altération des performances scolaires, seules étaient notées de discrètes anomalies cognitives révélées par le test de mémoire de travail verbale à court terme (données sur les nombres ou « digit Span »). Les enfants décrivaient, à travers des auto-questionnaires, plus d’anxiété et de manque de confiance en eux, troubles émotionnels non confirmés par leurs parents dans une publication des mêmes auteurs en 2008 (6) ; ces enfants étaient même décrits comme plus sociables que les contrôles non exposés à la DEX. En octobre 2015, lors du meeting de l’European Society for Pediatric Endocrinology à Barcelone, la même équipe suédoise a présenté, dans une communication orale présentée par L. Wallensteen, des résultats récents sur quelques enfants traités, atteints ou non, ne retrouvant aucune différence chez les garçons et une moins bonne performance des filles atteintes traitées ou non. Ainsi, les résultats suédois ne sont pas concordants et issus de petites cohortes d’enfants. À noter dans une étude polonaise, une meilleure performance des filles atteintes traitées (7). En parallèle, l’équipe américaine du Pr. MI New s’est penchée sur les aspects neurocognitifs avec un nombre beaucoup plus important d’enfants traités : en 2004, 174 mères traitées rapportaient l’absence de troubles neurocognitifs chez leurs enfants (8) et en 2012, les auteurs américains utilisant les mêmes tests neurocognitifs que les suédois, ont conclu à l’absence d’effets chez les enfants non atteints (9). Seule la vitesse de traitement de données était légèrement ralentie chez les filles atteintes traitées jusqu’à la fin de la grossesse. Concernant les possibles troubles métaboliques chez les enfants traités, un rapport a été donné par l’équipe du Pr. M.I New en 2010 lors d’une conférence internationale et les résultats publiés (10). Les auteurs ont conclu à l’absence d’anomalies métaboliques significatives à long terme chez les enfants traités (pas d’hypertension ni d’hyperglycémie). Des études multicentriques européennes et françaises sont en cours pour tenter de confirmer ces résultats chez l’enfant mais aussi chez l’adulte. Il est noté en contrepartie, en faveur du maintien du traitement par DEX chez les filles atteintes, que les études de suivi s’accordent sur l’impact psychologique traumatisant pour les parents d’une petite fille virilisée. De plus, l’hyperandrogénie pendant la vie fœtale serait décrite par certains auteurs comme responsable d’une altération du développement cérébral et de la masculinisation du comportement des filles après la naissance. Une étude récente réalisée par les Suédois sur une importante cohorte de filles et femmes atteintes rapportent une augmentation des troubles psychiatriques et de l’utilisation de substances (alcool, drogues), en lien possible avec d’une part l’hyperandrogénie et la diminution du cortisol pendant la vie fœtale et d’autre part l’impact de la virilisation et du devenir des petites filles atteintes, même opérées (11). Toutes les données publiées depuis ces dernières années ont conduit à la mise en place d’études multicentriques aux États-Unis, en Suède et en France. Un PHRC national va démarrer en France fin 2015, coordonné par notre laboratoire et associant les endocrino-pédiatres des centres de référence et de compétence français ainsi que des psychologues des centres de pédopsychiatrie. L’objectif principal est d’évaluer les effets neurocognitifs du traitement par DEX in utero chez les enfants non atteints, qui de fait ont reçu un excès de glucocorticoïdes durant la vie fœtale. Ce groupe sera comparé à un groupe témoin, composé d’enfants non atteints et non traités par DEX (fratrie d’enfants atteints et enfants scolarisés témoins). Les objectifs secondaires de cette étude permettront de déterminer les bénéfices du traitement par DEX chez les patients (notamment par diminution de l’hyperandrogénie cérébrale pendant la vie fœtale). Il faut souligner qu’en France plus de 400 enfants ont été traités in utero sans que soient notés à la naissance de malformations et de retard de croissance staturo-pondérale. |
||||||||
|
Références bibliographiques 1. David M, Forest MG. Prenatal treatment of congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. J Pediatr 1984;105(5):799-803. 2. Tardy-Guidollet V, Menassa R, Costa JM et al. New management strategy of pregnancies at risk of congenital adrenal hyperplasia using fetal sex determination in maternal serum: French cohort of 258 cases (2002-2011). Journal Clin Endocrinol Metab 2014;99:1180-8. 3. Miller WL. Fetal endocrine therapy for congenital adrenal hyperplasia should not be done. Best Pract Res Clin Endocrinol Metab. 2015;29(3):469-83. 4. Carmichael SL, Shaw GM, Ma C et al. Maternal corticosteroid use and orofacial clefts. American journal of obstetrics and gynecology 2007;197(6):585 :e1-7; discussion 683-4, e1-7. 5. Hirvikoski T, Nordenstrom A, Lindholm T et al. Cognitive functions in children at risk for congenital adrenal hyperplasia treated prenatally with dexamethasone. J Clin Endocrinol Metab 2007;92(2):542-8. 6. Hirvikoski T, Nordenstrom A, Lindholm T et al. Long-term follow-up of prenatally treated children at risk for congenital adrenal hyperplasia: does dexamethasone cause behavioural problems? Eur J Endocrinol 2008;159(3):309-16. 7. Maryniak A, Ginalska-Malinowska M, Bielawska A, Ondruch A. Cognitive and social function in girls with congenital adrenal hyperplasia — influence of prenatally administered dexamethasone. Child Neuropsychol 2012 ;20(1):60-70. 8. Meyer-Bahlburg HF, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Cognitive and motor development of children with and without congenital adrenal hyperplasia after early-prenatal dexamethasone. The Journal of clinical endocrinology and metabolism 2004;89(2):610-4. 9. Meyer-Bahlburg HF, Dolezal C, Haggerty R, Silverman M, New MI. Cognitive outcome of offspring from dexamethasone-treated pregnancies at risk for congenital adrenal hyperplasia due to 21-hydroxylase deficiency. European journal of endocrinology / European Federation of Endocrine Societies 2012;167(1):103-10. 10. New MI, Parsa AA. Long range outcome of prenatal treatment. Adv Exp Med Biol 2011;707:33-5. 11. Engberg H, Butwicka A, Nordenstrom A et al. Congenital adrenal hyperplasia and risk for psychiatric disorders in girls and women born between 1915 and 2010: A total population study. Psychoneuroendocrinology 2015;60:195-205. |
||||||||
 |
||||||||
|
Le contrôle hormonal est le principal facteur déterminant l’état de santé à l’âge adulte des patients avec une hyperplasie congénitale des surrénales par déficit en 21-hydroxylase Les hyperplasies congénitales des surrénales (HCS) sont des pathologies génétiques de transmission autosomique récessive qui se définissent par un déficit d’une des enzymes de la steroïdogenèse. Le déficit en 21-hydroxylase, en rapport avec des mutations du gène CYP21A2 est impliqué dans 90 à 95 % des HCS. Selon le degré de déficit enzymatique, il existe plusieurs formes cliniques allant de la forme classique (FC) à la forme non classique (FNC). Du fait de l’amélioration de la prise en charge des enfants présentant une HCS, se pose actuellement le problème du devenir de ces patients à l’âge adulte. Des études ont ainsi montré la présence de comorbidités chez ces patients à l’âge adulte (1, 2). Dans l’étude prospective multicentrique menée au Royaume-Uni sur 203 patients, l’indice de masse corporelle (IMC) était plus élevé que celui de la population générale, et l’existence d’une ostéopénie (40 %) ou d’une ostéoporose (7 %) était fréquente (3). Dans l’étude transversale américaine portant sur 244 patients CAH (183 classiques, 61 non classiques), l’obésité était présente chez environ un tiers des patients, et 37 % des adultes avaient une altération de la densité minérale osseuse (DMO) [4]. L’hirsutisme était commun chez les femmes (32 % classique ; 59 % des femmes NC) ainsi que la présence d’inclusion surrénaliennes intra-testiculaires chez les hommes (33 % des garçons ; 44 % des hommes). Ces études mettent en évidence que les patients adultes avec HCS ont des comorbidités associées à leur maladie. Cependant, ces complications ne concernent pas tous les patients adultes atteints d’HCS. Le rôle du traitement par glucocorticoïdes et la sévérité du génotype ont été proposés comme facteur prédisposant la survenue de ces complications. Récemment, N. Krone et al. ont réalisé une analyse phénotype/génotype chez 153 patients adultes (5). Ils n’ont trouvé aucune association entre le génotype et les paramètres cliniques définissant l’état de santé des patients. Les objectifs de notre étude, soutenue par un financement de l’association de patients « Association Surrénales », étaient de décrire la prévalence des comorbidités dans une population française de patients adultes atteints d’HCS et de chercher les facteurs déterminant (modifiables ou non) la survenue de ces comorbidités (6). Les critères étudiés ont été : IMC, ostéodensitométrie, cycles et hirsutisme chez les femmes, et présence d’inclusions testiculaires chez les hommes. Cent quatre patients (71 femmes, 33 hommes, 69 formes classiques et 35 formes non classiques), ont été analysés. Dans cette série, la présence d’un surpoids ou d’une obésité était retrouvée chez 44 % des patients, la présence de cycles menstruels irréguliers chez 50 % des femmes et une hyperandrogénie clinique chez 35 % des femmes. Un tiers des hommes présentaient des inclusions surrénaliennes intratesticulaires. Une altération de la DMO (ostéopénie ou ostéoporose) concernait 60 % de la cohorte. Les facteurs déterminants du surpoids ou de l’obésité, de la présence d’hirsutisme ou de cycles menstruels irréguliers étaient tous été liés au contrôle hormonal de la maladie, incluant les taux de 17-OH progestérone ou de ?4-androstènedione (p < 0,05). Le facteur déterminant d’une DMO anormale était un poids plus faible (p < 0,05). Le facteur déterminant de la présence d’inclusions testiculaires était la forme classique de la maladie (p = 0,002). La dose cumulée de glucocorticoïdes reçue au cours de la vie constituait un facteur de risque de surpoids ou obésité, et de la présence d’inclusions testiculaires (p = 0,05). La dose cumulée de fludrocortisone était un facteur de risque de présence d’inclusions (p = 0,03). Cette étude confirme la forte prévalence des complications à l’âge adulte des patients présentant une HCS diagnostiquée dans l’enfance, et le rôle clé du traitement et de l’équilibre hormonal. L’identification de ces facteurs doit permettre d’agir le plus précocement possible sur ces différents leviers afin d’optimiser le contrôle de la maladie. |
||||||||
|
Références bibliographiques 1. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao LM, Conway GS. Congenital adrenal hyperplasia in adults: a review of medical, surgical and psychological issues. Clin Endocrinol (Oxf) 2006;64(1):2-11. 2. Bachelot A, Plu-Bureau G, Thibaud E et al. Long-term Outcome of Patients with Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. Horm Res 2007;67(6):268-76. 3. Arlt W, Willis DS, Wild SH et al. ; United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE). Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab 2010;95(11):5110-21. 4. Finkielstain GP, Kim MS, Sinaii N et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2012;97(12):4429-38. 5. Krone N, Rose IT, Willis DS et al. ; United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE). Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab 2013;98(2):E346-354. 6. 6. Bachelot A, Golmard JL, Dulon J et al. Determination of risk factors for health outcomes in adult patients with childhood onset of congenital adrenal hyperplasia Eur J Endocrinol 2015;173(2):175-84. |
||||||||
|
Traitement chirurgical du Syndrome de Cushing par hyperplasie surrénalienne macronodulaire bilatérale : et si vous en laissiez une ? L’Hyperplasie surrénalienne macronodulaire bilatérale (HSMB) [cf. Newsletter surrénale n° 3] est une maladie génétique, liée dans 50 % des cas à une mutation du gène ARMC5 (1), qui se traduit par le développement très progressif d’une hyperplasie nodulaire, et d’une secrétion de cortisol autonome. Celle-ci peut évoluer vers un hypercortisolisme, et un syndrome de Cushing franc, généralement après 40 ans. Ce syndrome de Cushing surrénalien est indépendant de l’ACTH hypophysaire mais un de ses mécanismes est la secrétion d’ACTH par les cellules corticosurrénaliennes elles-mêmes ! (2). Jusqu’ici il était généralement admis que le traitement du syndrome de Cushing franc lié à l’HSMB était la surrénalectomie bilatérale. En effet les traitements médicaux étaient assez décevants, notamment en regard de l’espoir mis dans les traitements visant à inhiber l’activation des récepteurs ectopiques qui n’a été concrétisé que dans de très rares cas, et généralement de façon temporaire. Par ailleurs, il semble difficile de traiter les patients à vie par inhibiteurs de la stéroïdogénèse. Restait donc la chirurgie et dans les syndromes de Cushing franc le caractère bilatéral de la surrénalectomie semblait logique, sur la base de deux arguments, souvent implicites. Le premier est un argument « linéaire » : si le patient a un syndrome de Cushing franc avec un cortisol libre urinaire (CLU) à plus de deux fois la normale on ne doit pas pouvoir ramener ce CLU à la normale en enlevant une seule des deux surrénales. Le deuxième argument est « dynamique » : si on laisse une surrénale en place elle va continuer à grossir et si jamais le patient a été mis en rémission par une surrénalectomie unilatérale, il va alors forcément et rapidement récidiver sur sa surrénale restante. Cependant la surrénalectomie bilatérale a un prix élevé pour le patient : insuffisance surrénale totale et définitive, pathologie qui a elle-même une morbidité et mortalité non négligeables, même chez les patients bien éduqués. Cette insuffisance surrénale imposée est sans doute la raison pour laquelle certains endocrinologues de par le monde ont, malgré la logique apparente de la surrénalectomie bilatérale, adopté une attitude pragmatique : pourquoi ne pas essayer quand même d’enlever une seule surrénale ? En effet, si cela ne marchait pas il était toujours possible d’enlever la deuxième dans un deuxième temps chirurgical. Le prix à payer pour le patient étant alors de devoir subir deux interventions plutôt qu’une seule, ce qui semble un marché raisonnable pour une chance d’échapper à l’insuffisance surrénale. C’est ainsi qu’ont été rapportés des cas isolés, ou des petites séries de surrénalectomie unilatérale, avec des résultats étonnants : la surrénalectomie unilatérale marchait plutôt bien (3-8). Cependant ces publications ne sont pas arrivées à bousculer la surrénalectomie unilatérale, qui restait la référence. Pour essayer de mieux démontrer l’intérêt de la surrénalectomie unilatérale 4 centres français (Bordeaux, Grenoble, Kremlin-Bicêtre et Toulouse) ont alors décidé d’analyser en commun leurs 15 observations de surrénalectomie unilatérale chez des patients porteurs d’un syndrome de Cushing franc, ce qui représente la plus grosse série, avec un suivi post opératoire moyen de 5 ans (9). Les (nos !) résultats sont intéressants : 100 % des patients normalisent le CLU en post opératoire, avec un bénéfice clinique notamment sur l’indice de masse corporelle, la tension artérielle, la glycémie, et seulement 2 récidives, à 7 et 9 ans. Il y a même une surprise : 40 % des patients sont en insuffisance surrénale, dont la moitié récupèrent au cours du suivi. Comment est il possible qu’enlever une seule surrénale, certes la plus grosse, puisse être aussi efficace sur l’hypercortisolisme, et même parfois trop ? Pour cela il y a trois éléments de réponse. – L’argument linéaire est faux : le CLU n’est en fait pas du tout linéaire avec la production de cortisol : au-delà d’un certain taux de cortisol la CBG devient complètement saturée et le CLU augmente alors exponentiellement. Ainsi doubler la production de cortisol peut faire beaucoup plus que doubler le CLU. Inversement diviser par 2 la production de cortisol peut diviser le CLU par beaucoup plus que 2. – Il faut se rappeler que dans l’HSMB les cellules surrénaliennes sont sensibles à l’ACTH, mais après surrénalectomie unilatérale pour syndrome de Cushing franc l’ACTH hypophysaire, qui était freinée avant la chirurgie, va rester basse par inertie quel que soit le taux de cortisol. La surrénale restante ne va donc secréter que sa secrétion autonome, et si celle-ci est insuffisante elle ne pourra pas être »rattrapée » par une secrétion réactionnelle d’ACTH hypophysaire : il s’agit d’une insuffisance corticotrope, appelée à disparaître lorsque la secrétion d’ACTH recupérera de son inertie. – Il n’est pas très étonnant que la surrénale restante n’arrive pas à elle seule à assurer un hypercortisolisme : il faut se rappeler que l’HSMB est une pathologie congénitale de cinétique très lente, il faut plus de 40 ans pour que le patient arrive parfois à fabriquer trop de cortisol. Enlever une surrénale sur deux c’est faire faire au patient un voyage à remonter le temps, d’un temps de doublement du volume surrénalien, qui est certainement long. Ce « voyage » le ramène très probablement à un temps où sa masse surrénalienne, bien que déjà nettement supérieure à une surrénale normale n’était pas encore suffisante pour générer un hypercortisolisme franc, car on le rappelle, la stéroïdogènèse de ces surrénales est très inefficace. Bien sûr la surrénalectomie unilatérale n’est pas la panacée, il y aura des patients qui récidiveront, et le diagnostic de la transition entre normocortisolisme, hypercortisolisme infra-clinique et hypercortisolisme franc impose un suivi étroit. Mais une chose est sûre : une surrénalectomie unilatérale peut être transformée en surrénalectomie bilatérale. L’inverse n’est pas vrai ! |
||||||||
|
Référence bibliographique 1. Assié G, Libe R, Espiard S et al. J ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med 2013;369(22):2105-14. 2. Louiset E, Duparc C, Young J et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 2013;369(22):2115-5 3. Imohl M, Koditz R, Stachon A et al. [Catecholamine-dependent hereditary Cushing’s syndrome – follow-up after unilateral adrenalectomy]. Med Klin (Munich) 2002;97(12):747-53. 4. Lamas C, Alfaro JJ, Lucas T, Lecumberri B, Barcelo B, Estrada J. Is unilateral adrenalectomy an alternative treatment for ACTH-independent macronodular adrenal hyperplasia?: Long-term follow-up of four cases. European journal of endocrinology / European Federation of Endocrine Societies 2002;146(2):237-40. 5. Sato M, Soma M, Nakayama T et al. A case of adrenocorticotropin-independent bilateral adrenal macronodular hyperplasia (AIMAH) with primary hyperparathyroidism (PHPT). Endocr J 53(1):111-7. 6. Iacobone M, Albiger N, Scaroni C et al. The role of unilateral adrenalectomy in ACTH-independent macronodular adrenal hyperplasia (AIMAH). World J Surg 2008;32(5):882-9. 7. Mazzuco TL, Chaffanjon P, Martinie M, Sturm N, Chabre O. Adrenal Cushing’s syndrome due to bilateral macronodular adrenal hyperplasia: prediction of the efficacy of beta-blockade therapy and interest of unilateral adrenalectomy. Endocr J 2009;56(7):867-77. 8. Albiger NM, Ceccato F, Zilio M et al. An analysis of different therapeutic options in patients with Cushing’s syndrome due to bilateral macronodular adrenal hyperplasia: a single-centre experience. Clin Endocrinol (Oxf) 2015;82(6):808-15. 9. Debillon E, Velayoudom-Cephise FL, Salenave S et al. Unilateral Adrenalectomy as a first-line treatment of Cushing’s syndrome in patients with Primary Bilateral Macronodular Adrenal Hyperplasia. J Clin Endocrinol Metab jc20152662 [Epub ahead of print]. |
||||||||
 |
||||||||
|
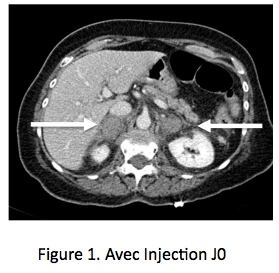
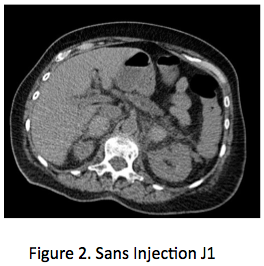
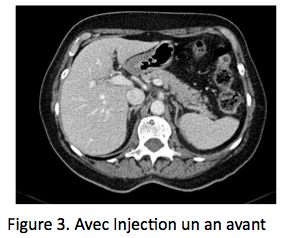
Le choc des images, le poids des dosages Une patiente de 70 ans est admise aux urgences pour douleur lombaire aiguë bilatérale prédominant à D, très intense. Dans ses antécédents on note un syndrome des anticorps anti-phospholipides, responsable il y a 2 ans d’une thrombose veineuse cérébrale justifiant un traitement anticoagulant oral, poursuivi depuis. La patiente présente rapidement des nausées et vomissements, une tension artérielle abaissée 90/60, pouls 100/mn. La natrémie est à 130 nmol/L, kaliémie 4,3 mmol/L, créatinine 60 umol/L et un scanner abdominal avec injection (figure 1) révèle des masses surrénaliennes bilatérales, 37 mm à droite et 34 mm à gauche, densité à 60 UH entourées d’une importante infiltration péri-surrénalienne. Il n’y a pas d’autres anomalies sur cette imagerie, qui fait évoquer aux urgentistes le diagnostic de nécrose hémorragique bilatérale des surrénales, avec insuffisance surrénale aiguë. Un traitement intraveineux par hydrocortisone, 100 mg bolus avec relais par 200 mg/24h et sérum salé isotonique est initié sans attendre, de même qu’un traitement antalgique par morphine. L’état hémodynamique s’améliore rapidement, de même que les douleurs lombaires, mais une suspicion de syndrome occlusif, attribué a posteriori à la morphine, fait réaliser le lendemain une deuxième TDM avec clichés sans injection (figure 2), qui retrouve une stabilité des masses surrénaliennes, dont la densité spontanée est identique à celle du premier scanner, démontrant ainsi que les masses surrénaliennes ont une densité spontanément élevée, sans prise de contraste. L’analyse des archives d’imagerie retrouve des clichés d’examen tomodensitométrique (TDM) réalisés un an plus tôt avec des surrénales normales (figure 3), l’ensemble confortant le diagnostic de nécrose hémorragique bilatérale des surrénales.
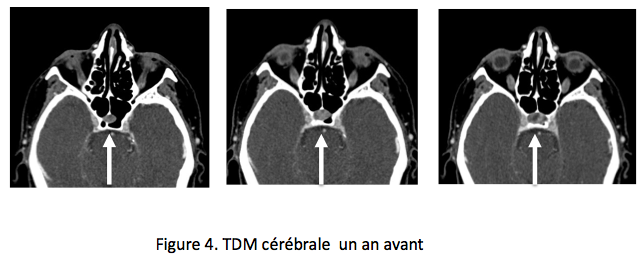
Jusqu’ici aucun dosage hormonal n’a été fait ! Deux semaines après, et alors que la posologie de l’hydrocortisone a été progressivement diminuée, un test au synacthène est réalisé, à 8 h du matin, 24 h après la dernière prise d’hydrocortisone. Sans surprise le cortisol est très bas (T0 58 nmol/L) et non stimulable (T60′ 59 nmol/L), en revanche, de façon inattendue, l’adrénocorticotrophine à T0 du synacthène est à peine élevée : 22 pmol/L (N 2,2-13,2), nettement plus basse qu’attendue pour cette insuffisance surrénalienne périphérique profonde. Un deuxième prélèvement réalisé deux semaines plus tard confirmera ce taux, qui fait évoquer une insuffisance hypophysaire. Une nouvelle exploration des archives d’imagerie de la patiente permet alors de retrouver des clichés de scanner cérébral réalisés un an auparavant pour l’exploration de sa thrombose veineuse cérébrale. Ils comprennent des clichés axiaux passant par le sinus sphénoïde et le plancher de la selle turcique, qui mettent en évidence une image arrondie dans le sinus sphénoïdal en continuité avec une image intrasellaire. L’endocrinologue se prend à imaginer un macroadénome hypophysaire qui aurait échappé jusqu’ici aux spécialistes de l’image (figure 4). Pas si vite : d’après eux il pourrait bien s’agir d’une selle turcique normale, peut-être un peu large… pas vraiment une image, peut-être un mirage ? Attendons l’IRM, et d’ailleurs l’exploration des autres axes hypophysaires ne retrouve pas de déficit…
Ainsi dans cette histoire une image se passe du dosage, puis le dosage appelle d’autres images ! Attention à ne pas nous laisser happer par la dialectique, restons cliniques. |
||||||||
 |
||||||||
|
MicroARNs et corticosurrénalome : de petites molécules associées à un grand risque L’incidence du corticosurrénalome (CS) est estimée entre 1 et 2 cas par million d’habitants par an. La survie à 5 ans est comprise entre 30 et 40 % mais elle est inférieure à 15 % au stade métastatique (1). La prise en charge thérapeutique repose essentiellement sur la résection chirurgicale. Les traitements médicaux pour les formes métastatiques du CS sont très limités. Il est donc crucial de mieux définir les mécanismes moléculaires impliqués dans le développement de ces tumeurs pour développer de nouvelles approches thérapeutiques. Plusieurs études réalisées ces dernières années ont permis d’établir une classification moléculaire des tumeurs corticosurrénaliennes bénignes et malignes, sur la base de leur profil d’expression génique analysé sur puces à ADN (2). Très récemment, une nouvelle étape décisive a été franchie grâce à la première étude de génomique intégrée du CS réalisée dans le cadre du réseau national COMETE (COrtico et MEdullosurrénale, Tumeurs Endocrines) et européen ENSAT (European Network for the Study of Adrenal Tumors) [3]. L’analyse de plus d’une centaine de CS par séquençage d’exome, transcriptome, miRNome, méthylome, et puces SNP (single nucleotide polymorphism) a permis l’identification de deux types moléculaires de corticosurrénalome, l’un associé à un bon pronostic après chirurgie et l’autre associé à un mauvais pronostic. Ces deux types de corticosurrénalomes correspondent à deux entités différentes caractérisées par un statut mutationnel, une méthylation des régions promotrices, des altérations chromosomiques et de l’expression génique ainsi qu’une signature microARNs spécifiques. Les microARNS sont une nouvelle classe de molécules découverte depuis une quinzaine d’années chez l’homme (4). Ce sont de petits ARN non-codants d’une vingtaine de nucléotides dont la fonction est de réguler négativement l’expression des gènes, essentiellement au niveau post-transcriptionnel. En s’appariant à la région 3’-non traduite de leurs ARN messagers (ARNm) cibles, ils provoquent la dégradation de ces ARNm ou bloquent leur traduction. Les microARNs sont impliqués dans de nombreux processus physiologiques tels que le développement, la croissance cellulaire, la prolifération et la différenciation, mais également pathologiques, tels que les maladies cardiovasculaires ou le cancer (5). Au cours du développement tumoral, les microARNs jouent un rôle de suppresseurs de tumeurs ou d’oncogènes selon le type et le contexte cellulaires. Comme pour les gènes « classiques », la dérégulation de leur expression dans le cancer est liée à des processus d’amplification, d’hyper-méthylation de promoteurs, de délétion, de mutations ou encore de translocations chromosomiques (6). Des signatures microARNs spécifiques ont été mises en évidence dans diverses tumeurs humaines. Fait très remarquable, l’expression de 217 microARNs a permis une classification de tumeurs peu différenciées meilleure que celle fondée sur l’expression de 16 000 ARNm (7). La puissance diagnostique de ces profils d’expression est probablement liée au fait qu’un seul microARN peut réguler l’expression de plusieurs gènes et être ainsi au cœur de plusieurs voies de signalisation altérées dans le cancer. Plus récemment, des microARNs circulants sous forme de complexes protéiques ou protégés dans des microvésicules appelés exosomes ont été identifiés dans différents fluides biologiques (sérum, plasma, salive, urine, etc.). Nombres d’études suggèrent un fort potentiel prédictif à ces molécules qui pourraient constituer une nouvelle catégorie de biomarqueurs très utiles en clinique (8). Contrairement aux avancées importantes réalisées pour les cancers à incidence élevée, la compréhension du rôle des microARNs dans la pathogenèse du corticosurrénalome n’en est qu’à ses prémices. À ce jour, huit études rétrospectives ont réalisé le profilage des microARNs dans les tumeurs corticosurrénaliennes. Des signatures sensiblement différentes ont été établies, probablement en raison de la faible taille et de l’hétérogénéité des cohortes ainsi que des méthodes d’analyse utilisées. Cependant, certains microARNs ont été trouvés dérégulés dans plusieurs études différentes, ce qui suggère leur rôle clé dans la pathogenèse du corticosurrénalome. Une perte de miR-195 et miR-335 a notamment été identifiée dans les corticosurrénalomes comparés aux adénomes ou à la corticosurrénale normale (9-12). À l’inverse, une surexpression de microARNs oncogéniques tels que miR-210 ou miR-483-5p a également été décrite (9-13). MiR-210 est un microARN critique dans la réponse cellulaire à l’hypoxie, une caractéristique commune de la plupart des tumeurs solides (14). MiR-483-5p suscite beaucoup d’intérêt car il est localisé dans le second intron du gène du facteur de croissance IFG2 soumis à empreinte parentale et fortement surexprimé dans près de 90 % des corticosurrénalomes (15). Le rôle oncogénique et pro-métastatique de miR-483-5p a récemment été décrit dans l’adénocarcinome du poumon ou son expression est activée par la voie de signalisation Wnt/?-Caténine (16). Bien que l’expression de miR-483-5p ait été trouvée corrélée à celle d’IGF2 dans le corticosurrénalome (11), il est possible que ce microARN ait un rôle spécifique dans le processus de tumorigenèse. Cette hypothèse est soutenue par l’observation que la seule surexpression de IFG2 ne conduit pas à la formation de tumeurs malignes de la corticosurrénale dans des modèles de souris transgéniques (17-18). Les niveaux d’expression de microARNs spécifiques ont également permis l’identification de sous-groupes de corticosurrénalomes associés à des survies globales différentes (10). En particulier, une forte expression de miR-483-5p et une faible expression de miR-195 sont associées à un mauvais pronostic (9). Dans une analyse visant à identifier des miRNAs dont l’expression permettrait de discriminer entre corticosurrénalome de bon pronostic et celui de mauvais pronostic, notre équipe a trouvé que 4 microARNs de la région Xq27 étaient plus exprimés et que 14 microARNs de la région 14q32 étaient moins exprimés dans les corticosurrénalomes de bon pronostic (12). Ces observations ont été confirmées récemment à plus grande échelle dans une cohorte multi-centrique européenne (3) : G. Assié et al. ont montré que le corticosurrénalome de bon pronostic est caractérisé par la surexpression de 11 microARNs de la région du chromosome Xq27 (groupe de microARNs miR 506-514), et la répression de 38 microARNs issus de la région chromosomique 14q32 soumise à empreinte parentale (groupe DLK1-MEG3). Cette étude de grande envergure représente une avancée importante dans la compréhension des mécanismes de l’oncogenèse corticosurrénalienne et ouvre des perspectives cliniques certaines en termes de prise en charge des patients. La première étude visant à évaluer la valeur diagnostique/pronostique de microARNs circulants pour le corticosurrénalome a montré qu’un niveau élevé de miR-483-5p et un niveau bas de miR-195 dans le sérum des patients porteurs de corticosurrénalomes sont des facteurs de mauvais pronostic (survie globale et survie sans récidive) [12]. L’augmentation de miR-483-5p circulant a été confirmée ultérieurement par deux autres études (19, 20). Cependant, bien que prometteurs, ces résultats ont été obtenus sur un nombre limité d’échantillons et restent à confirmer à plus grande échelle sur des cohortes indépendantes. Par ailleurs, une des limites de ces études rétrospectives est l’interférence potentielle des traitements avec les taux des microARNs circulants (mitotane, chimiothérapies). Des études prospectives fondées sur des critères d’inclusion précis et une méthodologie rigoureuse sont donc indispensables pour que les microARNs puissent être utilisés comme biomarqueurs pour le corticosurrénalome. Les applications potentielles des microARNs dépassent de loin le cadre de l’oncologie et touchent l’ensemble des pathologies humaines. Dans le corticosurrénalome, les microARNs permettraient d’envisager un diagnostic non-invasif par prélèvement sanguin ou même d’urine, sous réserve d’optimisation et de standardisation de leur détection dans les fluides biologiques. Enfin, les microARNs tumoraux peuvent devenir de nouvelles cibles thérapeutiques. Le défi majeur pour l’avenir est de comprendre comment les différents gènes cibles d’un microARN interagissent entre eux mais également comment un ensemble de microARNs coordonnent l’expression d’un même gène cible. La puissance de la bio-informatique et les analyses génomiques intégrées joueront un rôle déterminant dans ce défi plein de promesses. |
||||||||
|
Références bibliographiques 1. Libe R, Fratticci A, Bertherat, J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer 2007;14(1):13-28. 2. Assie G, Jouinot A, Bertherat, J. The ‘omics’ of adrenocortical tumours for personalized medicine. Nat Rev Endocrinol 2014;10(4):215-28. 3. Assie, G, Letouze E, Fassnacht et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 2014;46(6):607-12. 4. Lagos-Quintana M, Rauhut R, Lendeckel W et al. Identification of novel genes coding for small expressed RNAs. Science 2001;294(5543):853-8. 5. Calin, GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6(11):857-66. 6. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nature reviews. Cancer 2015;15(6):321-33. 7. Lu J, Getz G, Miska EA et al. MicroRNA expression profiles classify human cancers. Nature 2005;435(7043):834-8. 8. Wittmann J, Jack HM. Serum microRNAs as powerful cancer biomarkers. Biochim Biophys Acta 2010;1806(2):200-7. 9. Soon PS, Tacon LJ, Gill AJ et al. miR-195 and miR-483-5p Identified as Predictors of Poor Prognosis in Adrenocortical Cancer. Clin Cancer Res 2009;15(24):7684-92. 10. Ozata DM, Caramuta S, Velazquez-Fernandez D et al. The role of microRNA deregulation in the pathogenesis of adrenocortical carcinoma. Endocr Relat Cancer 2001;18(6):643-55. 11. Patterson EE, Holloway AK, Weng J et al. MicroRNA profiling of adrenocortical tumors reveals miR-483 as a marker of malignancy. Cancer 2011;117(8):1630-9. 12. Chabre, O, Libe R, Assie G et al. Serum miR-483-5p and miR-195 are predictive of recurrence risk in adrenocortical cancer patients. Endocr Relat Cancer 2013;20(4):579-94. 13. Tombol Z, Szabo PM, Molnar V et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer 16(3);895-906. 14. Chan YC, Banerjee J, Choi SY et al. miR-210: the master hypoxamir. Microcirculation 2012;19(3):215-23. 15. Gicquel C, Bertagna X, Schneid H et al. Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. The J Clin Endocrinol Metab 1994;78(6):1444-53. 16. Song Q, Xu Y, Yang C et al. (miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res 2014;74(11):3031-42. 17. Drelon C, Berthon A, Ragazzon B et al. Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One 2012;7(8):e44171. 18. Heaton JH, Wood MA, Kim AC et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and beta-catenin. American J Pathol 2012;181(3):1017-33. 19. Szabo DR, Luconi M, Szabo PM et al. Analysis of circulating microRNAs in adrenocortical tumors. Laboratory investigation; a journal of technical methods and pathology 2014:94(3):331-9. 20. Patel D, Boufraqech M, Jain M et al. MiR-34a and miR-483-5p are candidate serum biomarkers for adrenocortical tumors. Surgery 2013;154(6):1224-8;discussion 1229. |
||||||||