Item 244 – Adénome hypophysaire
________________________________________________________________________
Situations de départ
- 21 Asthénie.
- 25 Hypersudation.
- 26 Anomalies de la croissance staturo-pondérale.
- 33 Difficulté à procréer.
- 40 Écoulement mamelonnaire.
- 41 Gynécomastie.
- 42 Hypertension artérielle.
- 50 Malaise/perte de connaissance.
- 51 Obésité et surpoids.
- 57 Prise de poids.
- 61 Syndrome polyuro-polydypsique.
- 63 Troubles sexuels et troubles de l’érection.
- 78 Acné.
- 79 Hirsutisme.
- 80 Alopécie et chute des cheveux.
- 94 Troubles du cycle menstruel.
- 113 Puberté précoce ou retardée.
- 118 Céphalée.
- 138 Anomalie de la vision.
- 143 Diplopie.
- 148 Goitre ou nodule thyroïdien.
- 156 Ronflements.
- 164 Anomalie de l’examen clinique mammaire.
- 165 Palpitations.
- 182 Analyse de la bandelette urinaire.
- 194 Analyse du bilan thyroïdien.
- 202 Dysnatrémie.
- 208 Hyperglycémie.
- 226 Découverte d’une anomalie du cerveau à l’examen d’imagerie médicale.
- 251 Prescrire des corticoïdes par voie générale ou locale.
- 266 Consultation de suivi d’un patient polymédiqué.
- 279 Consultation de suivi d’une pathologie chronique.
- 281 Prescription médicamenteuse, consultation de suivi et éducation d’un patient diabétique de type 2 ou ayant un diabète secondaire.
- 282 Prescription médicamenteuse, consultation de suivi et éducation d’un patient hypertendu.
- 284 Consultation de suivi et éducation thérapeutique d’un patient avec hypothyroïdie.
- 306 Dépistage et prévention ostéoporose.
- 320 Prévention des maladies cardiovasculaires.
- 328 Annonce d’une maladie chronique.
- 352 Expliquer un traitement au patient (adulte, enfant, adolescent).
- 354 Évaluation de l’observance thérapeutique.
________________________________________________________________________________
________________________________________________________________________________
Hiérarchisation des connaissances
________________________________________________________________________________
| Rang | Rubrique | Intitulé | Descriptif |
| A | Diagnostic positif | Identifier les éléments d’un syndrome tumoral neurohypohysaire | Céphalées, anomalies du champ visuel, hypertension intracrânienne |
| A | Diagnostic positif | Identifier les signes cliniques orientant vers une hypersécrétion antéhypophysaire | Signes d’hyperprolactinémie, syndrome dysmorphique lié à l’excès de GH, syndrome de Cushing ou comorbidité révélatrice d’une hypersécrétion, comme l’apnée du sommeil, l’HTA |
| A | Diagnostic positif | Identifier un tableau d’insuffisance antéhypophysaire associée | Signes cliniques orientant vers une insuffisance corticotrope, thyréotrope, gonadotrope |
| A | Diagnostic positif | Connaître les signes d’un diabète insipide | Polyurie hypo-osmotique et polydipsie |
| B | Examens complémentaires | Savoir prescrire un bilan hormonal antéhypophysaire « statique » | Prolactine, IGF-1,cortisol à 8 h et ACTH à 8 h, FSH, LH, 17β-estradiol (F) ou testostérone (H) |
| B | Examens complémentaires | Connaître les indications de l’IRM hypophysaire en fonction du contexte clinique et biologique | Ne pas faire faire d’IRM systématique pour prolactinémie < 50 ng/ml |
| B | Diagnostic positif | Rechercher les autres causes d’hyperprolactinémie si un adénome à prolactine est suspecté | Connaître les hyperprolactinémies iatrogènes et fonctionnelles |
________________________________________________________________________________
Adénome hypophysaire
- Introduction
- Découverte de l’adénome hypophysaire devant un syndrome tumoral
- Découverte de l’adénome hypophysaire devant un syndrome d’hypersécrétion
- Découverte de l’adénome hypophysaire devant un tableau d’insuffisance antéhypophysaire
I Introduction
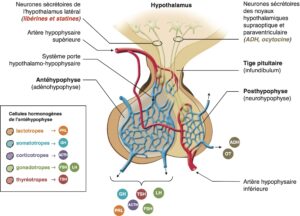
L’antéhypophyse est constituée de diverses populations cellulaires dont chacune produit de manière spécifique une hormone : les cellules lactotropes produisent la prolactine (PRL), les cellules somatotropes l’hormone de croissance (Growth Hormone, GH), les cellules corticotropes l’adrénocorticotropine (ACTH), les cellules gonadotropes l’hormone lutéinisante (LH) et la folliculostimuline (FSH), les cellules thyréotropes la thyréostimuline (TSH). Les hormones hypothalamiques (libérines et statines) qui régulent la sécrétion des hormones hypophysaires parviennent à l’antéhypophyse par le système porte hypothalamo-hypophysaire (fig. 1).
________________________________________________________________________________
Fig. 1
Anatomie fonctionnelle de l’hypophyse.
ACTH, Adrenocorticotrophic Hormone, hormone adrénocorticotrope ; ADH, Antidiuretic Hormone, vasopressine ; FSH, Follicle Stimulating Hormone, hormone folliculo-stimulante ; GH, Growth Hormone, hormone de croissance ; LH, Luteinizing Hormone, hormone lutéinisante, OT, ocytocine ; PRL, prolactine ; TSH, Thyroïd Stimulating Hormone, thyréostimuline.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Les adénomes hypophysaires sont des tumeurs bénignes développées aux dépens de l’hypophyse et qui, en fonction de leur taille et de leur caractère fonctionnel, sécrétant ou non, peuvent être responsables de trois grands types de signes :
- un syndrome tumoral hypophysaire, révélé par des troubles visuels (liés à la compression du chiasma optique situé quelques millimètres au-dessus de l’hypophyse) ou des céphalées, par un syndrome caverneux ou, plus fortuitement, à l’occasion d’une imagerie de la région hypothalamo-hypophysaire faite pour une raison indépendante (incidentalome hypophysaire) ;
- des syndromes d’hypersécrétion hormonale :
- hyperprolactinémie ;
- acromégalie secondaire à une hypersécrétion d’hormone de croissance ;
- hypercortisolisme(syndrome de Cushing) secondaire à une hypersécrétion d’ACTH stimulant la production surrénalienne de cortisol ;
- ou, plus rarement, hyperthyroïdie secondaire à une hypersécrétion de TSH par un adénome thyréotrope ;
- enfin, un syndrome d’insuffisance antéhypophysaire, portant généralement sur toutes les hormones hypophysaires (panhypopituitarisme).
En revanche, les adénomes hypophysaires ne s’accompagnent pas de diabète insipide.
Bien évidemment, ces trois grands cadres pathologiques ne s’excluent pas, ils sont même souvent associés.
II Découverte de l’adénome hypophysaire devant un syndrome tumoral
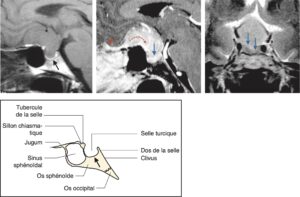
A Syndrome tumoral hypophysaire clinique
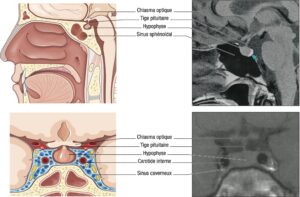
Il tient à la localisation de l’hypophyse et à ses rapports anatomiques, schématisés sur la fig. 2.
________________________________________________________________________________
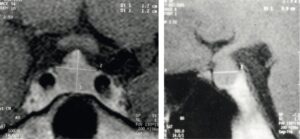
Fig. 2
Schémas anatomiques de profil et de face de la région hypophysaire, avec les coupes IRM sagittales et coronales en séquences pondérées T1 normales correspondantes.
(Source : CEEDMM, 2021, illustration du Pr Philippe Chanson.)
________________________________________________________________________________
1 Céphalées
Les céphalées sont typiquement rétro-orbitaires et localisées.
2 Troubles visuels
Il s’agit de troubles visuels par compression des voies optiques. Ils sont responsables d’une « gêne » visuelle, d’une impression de voile devant les yeux, d’une difficulté à fixer un point ou d’une sensation qu’il manque un mot à la lecture. L’acuité visuelle est le plus souvent normale du fait de l’absence d’atteinte du champ visuel central ; parfois, elle est diminuée en cas de lésion très volumineuse, négligée. L’examen du fond d’œil est normal le plus souvent.
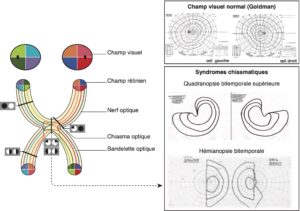
C’est l’atteinte du champ visuel, bien individualisée par l’étude du champ visuel à l’appareil de Goldman ou à la périmétrie automatisée, qui évoque le diagnostic devant une quadranopsie temporale supérieure ou, à un stade plus tardif, devant une hémianopsie bitemporale caractéristiques de la compression du chiasma optique (fig. 3).
________________________________________________________________________________
Fig. 3
Schémas des voies optiques.
Représentation d’un champ visuel à l’appareil de Goldman : champ visuel normal (en haut), d’une quadranopsie bitemporale supérieure (au milieu), d’une hémianopsie bitemporale (en bas) en relation avec une compression chiasmatique.
(Source : CEEDMM, 2021, illustration du Pr Gérald Raverot.)
________________________________________________________________________________
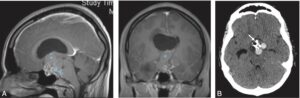
3 Tableau d’apoplexie hypophysaire.
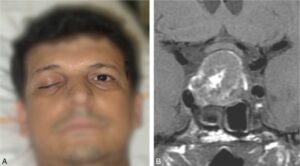
Plus rarement, on observe un tableau d’apoplexie hypophysaire à début brutal : céphalées violentes, photophobie (pseudo-syndrome méningé), fréquemment paralysie oculomotrice (par compression des nerfs crâniens passant dans le sinus caverneux) avec ptosis et diplopie (fig. 4), syndrome confusionnel, voire coma, troubles visuels par compression chiasmatique aiguë. S’y associent des signes d’insuffisance hypophysaire et notamment corticotrope aigus. L’apoplexie peut révéler un adénome méconnu et le tableau évoque une urgence neurochirurgicale. L’imagerie faite en urgence permet le diagnostic en montrant un adénome en voie de nécrose ou d’hémorragie.
________________________________________________________________________________
Fig. 4
Ptosis (A) en rapport avec une atteinte oculomotrice par atteinte du III, dans le cadre d’une apoplexie hypophysaire, bien visible sur l’IRM (B) qui met en évidence un macroadénome hypophysaire à développement suprasellaire avec des zones de nécrose et d’hémorragie.
(Clichés dus à l’obligeance du Pr J.-F. Bonneville.)
________________________________________________________________________________
B Imagerie tumorale hypophysaire : IRM
C’est maintenant l’imagerie par résonance magnétique (IRM) qui est l’examen de référence de la région hypothalamo-hypophysaire. L’aspect normal de l’hypophyse est indiqué sur la figure 15.2. Le scanner de la région hypophysaire n’est utilisé qu’en cas de contre-indication à l’IRM.
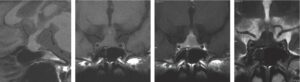
1 Microadénomes
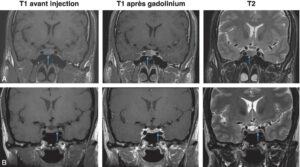
Les microadénomes, définis par leur taille inférieure à 10 mm de diamètre, apparaissent sous la forme d’une image arrondie, homogène (fig. 5).
________________________________________________________________________________
Fig. 5
Microadénomes hypophysaires vus par IRM. Coupes coronales en pondération T1 avant injection, T1 après injection de gadolinium, T2.
A. Microadénome latéralisé à droite (flèches) non visible en T1 (isosignal), apparaissant en hyposignal par rapport à l’hypophyse normale injectée, et en hyposignal T2. Noter aussi le déplacement de la tige pituitaire vers la gauche. B. Microadénome latéralisé à gauche (flèches) non visible en T1 (isosignal), non visible après injection, et visible uniquement en hypersignal T2.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Après injection, le microadénome apparaît hypointense au reste de l’hypophyse du fait d’un retard de prise de contraste par rapport à l’hypophyse saine qui prend le contraste de façon homogène en T1. Ces microadénomes peuvent augmenter le volume global de l’hypophyse, faire bomber son bord supérieur qui apparaît alors convexe, faire dévier latéralement la tige pituitaire dans le sens opposé de la lésion (signes indirects).
2 Macroadénomes
Les macroadénomes sont définis par leur taille supérieure à 10 mm de diamètre.
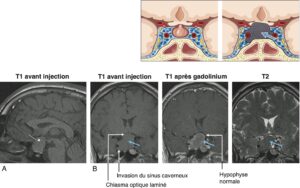
Ils apparaissent généralement isointenses au reste du parenchyme cérébral en T1 avant injection, hypointenses par rapport à l’hypophyse saine et hyperintenses par rapport au reste du parenchyme cérébral après injection (fig. 6).
________________________________________________________________________________
Fig. 6
Macroadénome hypophysaire vu par IRM.
A. Coupe sagittale : T1 avant injection. B. Coupes coronales : T1 avant injection ; T1 après injection de gadolinium, T2.
L’adénome (flèche bleue) apparaît isointense au parenchyme cérébral en T1 ; après injection de gadolinium, il apparaît hypointense par rapport à l’hypophyse saine, comprimée par l’adénome et refoulée vers la gauche et plutôt hyperintense par rapport au parenchyme cérébral ; en T2, l’adénome est iso-/hypointense par rapport au parenchyme cérébral. Le chiasma optique est refoulé, laminé par l’expansion suprasellaire de l’adénome. L’adénome envahit le sinus caverneux droit.
(Source : CEEDMM, 2021, et illustration du Pr Philippe Chanson.)
________________________________________________________________________________
On étudie l’expansion suprasellaire éventuelle en haut vers la citerne optochiasmatique — contact éventuel avec le chiasma optique qui peut être comprimé, refoulé (fig. 6) voire laminé, ou avec les bandelettes optiques, voire les nerfs optiques — et celle éventuelle vers le troisième ventricule. On analyse l’extension inférieure vers le sinus sphénoïdal et l’expansion latérale voire l’invasion du sinus caverneux (fig. 6).
3 Diagnostic différentiel en imagerie
Craniopharyngiome intrasellaire (fig. 7)
Le plus souvent en position suprasellaire, il peut également être intrasellaire. Il apparaît souvent sous la forme d’une masse hétérogène à composantes multiples : tissulaire, kystique, hémorragique. En T1, il est en hypo- ou en hypersignal, en T2 en hypersignal souvent associé à un hyposignal. Les calcifications ne sont pas vues en IRM mais sont bien visibles au scanner (clichés sans injection en fenêtre osseuse).
________________________________________________________________________________
Fig. 7
Craniopharyngiome intra- et suprasellaire.
Le craniopharyngiome apparaît à l’IRM (A) sous la forme d’une masse hétérogène (flèche bleue) à composantes multiples : tissulaire, kystique, hémorragique. Un kyste volumineux (indiqué par une étoile) s’est développé en suprasellaire dans le troisième ventricule et vient obstruer le foramen interventriculaire (trou de Monro) à droite, entraînant une dilatation du ventricule latéral droit et une hypertension intracrânienne. Les calcifications ne sont pas vues en IRM mais sont bien visibles au scanner sur les clichés sans injection en fenêtre osseuse (flèche blanche) (B).
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Méningiome intrasellaire
Une condensation anormale de l’os en regard de la lésion est bien visible au scanner. La prise de contraste est intense en IRM (fig. 8). La dure-mère voisine de la tumeur est souvent épaissie et prend de façon très intense le contraste. L’aspect spiculé de la dure-mère accolée à la lésion est caractéristique.
________________________________________________________________________________
Fig. 8
Méningiome suprasellaire à développement intrasellaire.
La lésion intrasellaire présente une expansion suprasellaire mais certaines caractéristiques permettent de la différencier d’un adénome hypophysaire : la selle turcique n’est pas déformée (flèche noire) ; l’hypophyse normale (indiquée par les flèches bleues) est au fond de la selle turcique sous la lésion. En fait il s’agit d’un méningiome qui s’implante sur le jugum sphénoïdal, l’infiltration durale y donnant un aspect caractéristique en « queue de comète » (tête de flèche rouge), et qui descend dans la selle turcique (flèche rouge pointillée), refoulant l’hypophyse normale vers le bas.
________________________________________________________________________________
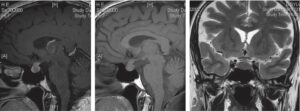
Grosse hypophyse de la femme jeune
Chez l’adolescente ou la femme jeune, à l’occasion généralement d’une IRM faite pour une autre raison, on découvre parfois de manière fortuite, une hypophyse un peu bombée vers le haut, qui peut en imposer pour un adénome à expansion suprasellaire, d’autant plus que la selle turcique est peu profonde (fig. 9).
________________________________________________________________________________
Fig. 9
Grosse hypophyse de la femme jeune.
L’hypophyse normale de cette jeune femme bombe vers le haut et « sort » de la selle turcique qui est plate et étroite. L’aspect pourrait en première analyse faire suspecter un adénome à expansion suprasellaire mais, à la différence des adénomes, le tissu hypophysaire est homogène et se rehausse de manière homogène avec le produit de contraste.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Autres tumeurs ou infiltrations
Métastases (fig. 10), sarcoïdose, histiocytose, tuberculose, hypophysite auto-immune (fig. 11), etc.
________________________________________________________________________________
Fig. 10
Métastase hypophysaire.
La lésion, fréquemment intra- et suprasellaire, est souvent à l’origine de céphalées et d’un diabète insipide. Généralement le cancer (cancer du sein, cancer bronchopulmonaire…) est connu. Dans le contexte, le diagnostic est évident.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
________________________________________________________________________________
Fig. 11
Hypophysite auto-immune.
La lésion apparaissant souvent en fin de grossesse ou dans le post-partum, comme c’est le cas chez cette patiente, est fréquemment révélée par des céphalées et des signes d’insuffisance hypophysaire (et parfois un diabète insipide). La selle turcique non déformée, l’infiltration de la totalité de la glande (on ne visualise pas l’hypophyse normale), la forte prise de contraste après injection, le contexte permettent de la différencier de l’adénome hypophysaire.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Contrairement aux adénomes hypophysaires, toutes ces lésions peuvent être responsables d’un diabète insipide central défini par une polyurie (3 litres par 24 heures) d’urines hypotoniques (osmolarité urinaire < 300 mOsm/l) accompagnée d’une polydipsie équivalente à la diurèse, de manière à maintenir une osmolalité plasmatique normale. En présence d’un syndrome polyuro-polydipsique, la notion d’une lésion hypothalamo-hypophysaire à l’IRM rend inutile la réalisation d’un test de restriction hydrique : le diagnostic de diabète insipide central par carence en ADH (ou arginine-vasopressine) est évident.
On rappelle que le test de restriction hydrique, qui doit être fait en milieu hospitalier car il est dangereux, n’est indiqué qu’en cas d’absence de lésion hypothalamo-hypophysaire, pour différencier un diabète insipide central idiopathique (souvent séquellaire d’une neuro-infundibulite passée inaperçue) d’une polydipsie primaire par trouble primitif de la soif.
III Découverte de l’adénome hypophysaire devant un syndrome d’hypersécrétion
A Hyperprolactinémie
L’hyperprolactinémie est une pathologie fréquente (1 à 1,5 % des adultes). La découverte d’une hyperprolactinémie fait chercher (et parfois trouver) un adénome hypophysaire — qu’il s’agisse d’un adénome à prolactine ou qu’il s’agisse d’un adénome d’une autre nature, responsable d’une hyperprolactinémie dite de déconnexion.
Mais n’oublions pas que, dans la majorité des cas, l’hyperprolactinémie est d’origine médicamenteuse.
1 Signes amenant à chercher une hyperprolactinémie
Chez la femme
Galactorrhée
La galactorrhée est spontanée ou plus souvent uniquement provoquée (dans 80 % des cas). Elle n’est significative que si elle est faite de liquide lactescent et qu’elle survient à distance du post-partum. La découverte d’une galactorrhée n’est pas synonyme d’hyperprolactinémie : la grande majorité des femmes consultant pour une galactorrhée ont même une prolactinémie normale. Toutefois, toute galactorrhée impose un dosage de prolactine (10 % des femmes hyperprolactinémiques ont une galactorrhée isolée sans troubles des règles).
Perturbations du cycle menstruel ou infertilité
L’aménorrhée est le signe le plus fréquent : près de 90 % des femmes ayant une hyperprolactinémie ont une absence totale de règles (aménorrhée) ou une oligoménorrhée (moins de quatre cycles par an) et, dans la moitié des cas, des irrégularités menstruelles ou un allongement progressif des cycles (spanioménorrhée) avaient été notés dans les mois ou les années précédents.
L’hyperprolactinémie inhibe la sécrétion pulsatile de GnRH hypothalamique et, par voie de conséquence, la sécrétion de LH et de FSH (hypogonadisme hypogonadotrope), et perturbe l’ovulation et le développement du corps jaune. On note souvent une baisse de la libido et parfois une dyspareunie liée à la sécheresse vaginale qui indique un effondrement de l’œstradiol.
Parfois, les règles sont bien régulières et le tableau clinique se limite à une anovulation (5 % des cas environ) avec une courbe de température plate, une absence de sécrétion de progestérone et une infertilité (20 % des infertilités d’origine hormonale sont liées à une pathologie de la prolactine). Tout trouble des règles à type d’aménorrhée ou d’oligoaménorrhée, toute infertilité justifient donc un dosage plasmatique de la prolactine (cf. Item 38 – Infertilité du couple et Item 42 – Aménorrhée).
Chez l’homme
L’hyperprolactinémie peut, rarement, provoquer une galactorrhée ou une gynécomastie — la glande mammaire a besoin d’œstrogènes pour se développer. Plus fréquemment, elle est à l’origine de troubles sexuels : baisse de la libido, voire troubles de l’érection. En fait, ces signes sont souvent inavoués ou négligés par le patient ou son médecin et, si c’est une tumeur volumineuse qui est à l’origine de l’hyperprolactinémie, c’est plus souvent un syndrome tumoral (troubles visuels par compression du chiasma optique, céphalées, etc.) ou encore un panhypopituitarisme qui amènent à suspecter le diagnostic. Quoi qu’il en soit, l’exploration d’un hypogonadisme chez l’homme impose un dosage de prolactine (+++).
Dans les deux sexes
À long terme, la persistance d’une hyperprolactinémie, du fait des conséquences de l’hypogonadisme, est responsable d’une déminéralisation osseuse et d’un risque d’ostéoporose.
2 Stratégie diagnostique devant une hyperprolactinémie
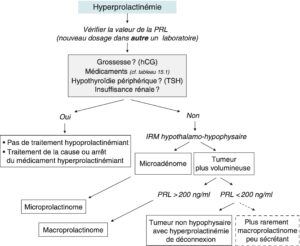
L’hyperprolactinémie, lorsqu’elle dépasse 20 ng/ml chez l’homme comme chez la femme, impose une stratégie diagnostique assez stéréotypée, résumée dans la figure 12.
________________________________________________________________________________
Fig. 12
Stratégie diagnostique devant une hyperprolactinémie.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
1re étape – Vérifier la réalité de l’hyperprolactinémie
Un contrôle de la prolactinémie dans un laboratoire d’hormonologie spécialisé est indispensable. En effet, des fausses hyperprolactinémies sont souvent observées, liées aux kits de dosage utilisés dans certains laboratoires non spécialisés.
Parfois, alors même qu’il n’existe aucun symptôme en rapport avec une hyperprolactinémie, le dosage de prolactine (réalisé de façon « systématique ») trouve une valeur élevée, en rapport avec une « macroprolactinémie » (à ne pas confondre avec le macroprolactinome), c’est-à-dire des agrégats de prolactine perturbant le dosage et donnant ce résultat de fausse hyperprolactinémie. La variabilité des résultats du dosage d’une trousse à l’autre est évocatrice. La chromatographie de la prolactine, en séparant la prolactine monomérique de la prolactine présente sous forme polymérique (agrégats de prolactine par des immunoglobulines), permet de corriger l’erreur de dosage — on ne doit tenir compte que de la prolactine monomérique.
2e étape – Éliminer les hyperprolactinémies de causes générales et médicamenteuses
La grossesse (marqueur hCG), l’hypothyroïdie périphérique (marqueur TSH, cause classique mais très rare d’hyperprolactinémie) et l’insuffisance rénale chronique sont facilement écartées.
Un interrogatoire soigneux permet enfin de s’assurer de l’absence de prise médicamenteuse susceptible d’élever la prolactinémie (fig. 13, tableau 1).
Fait essentiel, l’hyperprolactinémie secondaire est généralement < 150 ng/ml et n’atteint des valeurs très élevées (200 voire 350 ng/ml) qu’en cas de traitement par neuroleptiques et antiémétiques (dompéridone, sulpiride).
________________________________________________________________________________
Tableau 1
Principales causes médicamenteuses d’hyperprolactinémie. (Source : CEEDMM, 2021.)
| – Neuroleptiques (phénothiazines, halopéridol, sulpiride)
– Antidépresseurs (tricycliques et IMAO) – Métoclopramide, dompéridone – Œstrogènes – Morphiniques – Vérapamil – Méthyldopa |
________________________________________________________________________________
________________________________________________________________________________
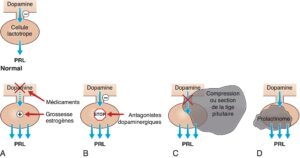
Fig. 13
Physiopathologie des différentes étiologies d’hyperprolactinémie. Normalement la sécrétion de PRL par la cellule lactotrope est freinée en permanence par la dopamine hypothalamique. Une hyperprolactinémie peut être observée dans diverses situations physiologiques et pathologiques.
A. La grossesse et les fortes doses d’œstrogènes d’une part, freinent la sécrétion de dopamine et, d’autre part, stimulent directement la production de PRL par la cellule lactotrope normale. B. Les antagonistes du récepteur de la dopamine (neuroleptiques +++) s’opposent aux effets de la dopamine sur la cellule lactotrope normale, ce qui défreine la production de PRL. C. L’hyperprolactinémie de déconnexionest liée à une interruption du trafic de la dopamine dans la tige pituitaire(section de la tige pituitaire ou compression par une tumeur de la région hypothalamo-hypophysaire) : la cellule lactotrope normale est défreinée, ce qui augmente la production de PRL. D. Le prolactinome est une tumeur développée aux dépens des cellules lactotropes, qui s’accompagne d’une production excessive de PRL. (PRL : prolactine).
(Source : CEEDMM, 2021, illustration du Pr Philippe Chanson, adapté de : Chanson P. Hyperprolactinémie. EMC, Traité de Médecine Akos. 2018 ; 13(2) : 1–6, Article 1-1300.)
________________________________________________________________________________
3e étape – Trouver la lésion hypothalamo-hypophysaire responsable
Lorsque les causes médicamenteuses ou générales sont éliminées, il faut envisager la possibilité d’une tumeur de la région hypothalamo-hypophysaire. Pour cela, une IRM est indiquée pour identifier des lésions tumorales, de taille très variable (cf. supra). Lorsque l’IRM n’est pas disponible ou contre-indiquée, on peut se contenter d’un scanner hypophysaire.
Il peut s’agir d’un microadénome intrasellaire (diamètre < 10 mm) souvent intra-hypophysaire ; la prolactinémie est alors modérément augmentée, entre 30 et 100 ng/ml.
À l’opposé, l’examen neuroradiologique peut révéler une volumineuse tumeur de la région hypophysaire, comprimant parfois le chiasma optique (et imposant alors la réalisation urgente d’un examen du champ visuel et la mesure de l’acuité visuelle). Cette volumineuse tumeur peut correspondre à :
- un macroadénome à prolactine, ou macroprolactinome ;
- une tumeur d’une autre origine, non prolactinique (à point de départ hypophysaire ou hypothalamique), associée à une hyperprolactinémie de déconnexion hypothalamo-hypophysaire(cf. 15.13).
La distinction entre ces deux étiologies est difficile.
En cas de tumeur non prolactinique avec hyperprolactinémie de déconnexion, la prolactinémie est très rarement supérieure à 150–200 ng/ml.
En cas de prolactinome, la prolactine est fonction de la masse tumorale — si > 150–200 ng/ml, il s’agit quasi obligatoirement d’un prolactinome ; mais si < 150–200 ng/ml, ce peut être un prolactinome peu sécrétant mais c’est plus généralement une tumeur non prolactinique.
L’évolution de la masse tumorale (et non pas de la prolactinémie) sous agoniste dopaminergique peut aider à faire la distinction : la lésion diminuera de taille si c’est un prolactinome et restera de taille identique si c’est une tumeur non prolactinique. Cette distinction est indispensable car dans le second cas une prise en charge chirurgicale est généralement nécessaire… alors qu’en cas de macroprolactinome, c’est plutôt le traitement médicamenteux (agonistes dopaminergiques) qui sera choisi.
L’évaluation des autres fonctions hypophysaires est indispensable en cas de lésion hypophysaire tumorale (cf. infra).
B Acromégalie (excès d’hormone de croissance, GH)
1 Syndrome dysmorphique et diagnostic
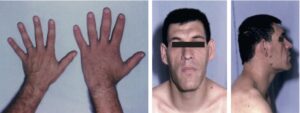
Le syndrome dysmorphique de l’acromégalie peut amener à évoquer le diagnostic (fig. 14).
________________________________________________________________________________
Fig. 14
Acromégalie.
Mains d’un homme acromégale (noter la comparaison avec la main d’un adulte normal ; le patient a dû faire élargir sa bague à deux reprises). Syndrome dysmorphique de l’acromégalie.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Les extrémités (mains, pieds) sont élargies, les doigts sont élargis, épaissis, boudinés et la peau de la paume des mains et de la plante des pieds est épaissie. Le patient a dû faire élargir bague ou alliance et a changé de pointure. Le visage est caractéristique : le nez est élargi, épaissi. Les pommettes sont saillantes, le front bombé, les lèvres épaisses, les rides sont marquées. Il existe une tendance au prognathisme. La comparaison avec des photographies antérieures met en évidence la transformation lente, insidieuse sur plusieurs années (à l’anamnèse, les premiers troubles de la maladie remontent généralement à 5 à 10 ans auparavant), ce qui explique que l’entourage ou le médecin traitant n’aient rien remarqué.
Si l’acromégalie est ancienne, les déformations peuvent aussi toucher le reste du squelette : cyphose dorsale, sternum projeté en avant, voire aspect exceptionnel du polichinelle.
2 Signes fonctionnels et généraux
Au syndrome dysmorphique s’associent des signes fonctionnels et généraux, tels que :
- sueurs, surtout nocturnes, malodorantes ;
- céphalées(que l’adénome hypophysaire en cause soit volumineux ou non) ;
- paresthésies des mains, voire authentique syndrome du canal carpien ;
- douleurs articulaires pouvant conduire à consulter ;
- asthénie fréquente ; parfois syndrome dépressif ;
- une HTA, trouvée chez près d’un acromégale sur deux.
Le patient (ou surtout son entourage) se plaint parfois d’un ronflement nocturne et l’interrogatoire de l’entourage amène parfois à évoquer un authentique syndrome d’apnées du sommeil, avec pauses respiratoires nocturnes et endormissement diurne, qu’il faut authentifier par une polysomnographie.
Ces symptômes sont non spécifiques et leur progression lente explique le retard au diagnostic classique. Il faut donc envisager le diagnostic et faire un dépistage biologique lorsqu’un patient présente au moins deux comorbidités telles qu’asthénie, prise pondérale, sueurs, ronflement (SAS), syndrome du canal carpien, diabète de type 2, arthralgies diffuses.
3 Complications de l’acromégalie
Les complications de l’acromégalie peuvent révéler la maladie et doivent être recherchées.
Complications cardiovasculaires
Observation fréquente d’une hypertrophie myocardique (septum et paroi postérieure du ventricule gauche) à l’échographie, avec parfois un simple dysfonctionnement diastolique (trouble de la compliance) et un débit cardiaque basal augmenté (syndrome hyperkinétique). Si l’atteinte cardiaque évolue, un tableau d’insuffisance cardiaque congestive se constitue, responsable de signes fonctionnels survenant d’abord à l’effort puis permanents. Les complications cardiovasculaires sont la première cause de mortalité des acromégales.
Arthropathie acromégalique périphérique
Elle touche typiquement les grosses articulations : genoux, épaules, mains, poignets et hanche. Les arthralgies sont de rythme mécanique mais aussi parfois inflammatoire. À la radiographie, les interlignes articulaires sont élargis ; on note la présence d’ostéophytes exubérants, d’ossifications des insertions tendineuses.
Le rhumatisme acromégalique touche surtout le rachis : lombalgies de type mécanique le plus souvent.
À la radiographie, on observe la classique spondylose d’Erdheim (coulées ostéophytiques antérieures et latérales des corps vertébraux, aspect biconcave des vertèbres et concavité exagérée du mur vertébral postérieur).
Diabète ou intolérance au glucose
Le diabète ou l’intolérance au glucose sont fréquents.
Syndrome d’apnées du sommeil
Ce syndrome est présent chez plus des deux tiers des malades. Les apnées sont obstructives ou mixtes.
Autres
Organomégalie (hépatomégalie, splénomégalie, etc.). Les goitres, souvent multinodulaires, sont fréquents.
Des polypes du côlon sont trouvés de façon plus fréquente (coloscopie régulière).
4 Diagnostic biologique de l’acromégalie
- Le test de dépistage qui permet le diagnostic en cas de suspicion clinique est le dosage d’IGF-1. Il doit être interprété en fonction de l’âge.
- Le dosage isolé de la GH:
- il n’a pas de valeur diagnostique car la sécrétion de GH chez le sujet normal est variable dans le nycthémère ;
- la concentration de GH est tantôt basse, indétectable, tantôt (et cela de façon brève, durant quelques minutes) très élevée (le pic pouvant atteindre 10 à 20 ng/ml).
- Le diagnostic :
- repose donc sur la mise en évidence d’une absence de freinage de la GH lors de l’hyperglycémie provoquée par voie orale (HGPO) ;
- en effet :
- chez un sujet normal, la GH s’abaisse toujours au-dessous de 0,4 ng/ml (1 mUI/l) après HGPO, alors qu’elle reste supérieure à 0,4 ng/ml chez l’acromégale ;
- parfois une réponse paradoxale de la GH (stimulation) est présente chez l’acromégale.
5 Nécessité du triple bilan
Une fois le diagnostic établi, un triple bilan est nécessaire :
- un bilan tumoral, afin de mettre en évidence si l’adénome responsable de l’hypersécrétion est un microadénome ou un macroadénome (cf. supra) ;
- un bilan du retentissement fonctionnel hypophysaire de la tumeur, afin de vérifier si, en plus de l’hypersécrétion de GH, l’adénome n’est pas responsable d’une altération des autres fonctions hypophysaires (cf. infra) et d’une cosécrétion fréquente de prolactine ;
- un bilan du retentissement de l’acromégalie, à la recherche des complications de la maladie.
C Hypercortisolisme (ou syndrome de Cushing)
Le syndrome de Cushing est l’ensemble des manifestations cliniques et biologiques engendrées par un excès chronique de glucocorticoïdes. En dehors des causes iatrogènes (corticothérapie), les adénomes hypophysaires corticotropes (maladie de Cushing) sont la cause la plus fréquente du syndrome de Cushing.
1 Tableau clinique et diagnostic
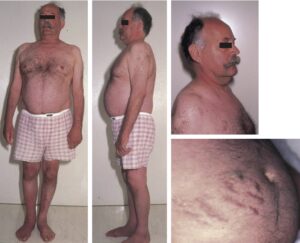
Le tableau clinique fait évoquer le diagnostic (pour exemple : fig. 15 ; cf. aussi Item 224 – Hypertension artérielle de l’adulte).
________________________________________________________________________________
Fig. 15
Patient atteint d’un syndrome de Cushing ACTH-dépendant.
Noter l’obésité faciotronculaire, les vergetures pourpres abdominales et l’amyotrophie des quadriceps.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
Anomalies morphologiques
Ces anomalies sont acquises (comparaison avec des photographies antérieures).
Signes spécifiques
Les signes spécifiques sont les signes secondaires à l’effet catabolique et antianabolique des glucocorticoïdes sur le métabolisme protidique. L’amyotrophie prédomine au niveau des ceintures et de l’abdomen et peut être responsable d’une fatigabilité à la marche. Elle est parfois discrète (manœuvre du tabouret). L’atrophie cutanée et sous-cutanée est responsable d’une lenteur à la cicatrisation.
La peau (dos des mains) est amincie (en « feuille de papier à cigarette »). Des ecchymoses surviennent au moindre choc. Les vergetures cutanées sont larges (> 1 cm), pourpres, orientées horizontalement sur les flancs et à la racine des membres, ou à disposition radiaire dans la région mammaire et péri-ombilicale. La peau du visage est érythrosique, congestive avec varicosités et télangiectasies.
Signes moins spécifiques
La prise pondérale est modérée, généralement d’une dizaine de kilogrammes, et présente une topographie particulière, faciotronculaire (modification de la répartition des graisses), respectant les extrémités. Le visage devient arrondi, bouffi, avec une hypertrophie des boules de Bichat. On note un comblement des creux sus-claviculaires et un aspect en « bosse de bison » au niveau de la nuque, ainsi qu’une augmentation du rapport taille/hanche. Enfin, cette obésité contraste avec une amyotrophie des membres.
Autres anomalies morphologiques
Elles sont moins spécifiques :
- les symptômes d’hyperandrogénie:
- ils se limitent généralement à un hirsutisme modéré (duvet de la lèvre supérieure, poils fins parsemés au niveau du menton, ébauche de favoris) et à une séborrhée du visage et du cuir chevelu avec des lésions acnéiques ;
- ces dernières ne tiennent pas au cortisol (qui est dénué d’effet androgénique) mais à diverses causes du syndrome de Cushing qui peuvent s’associer à une sécrétion d’androgènes surrénaliens ou ovariens (SOPK secondaire) ;
- des œdèmes des membres inférieurs sont parfois notés.
2 Autres manifestations cliniques
D’autres manifestations cliniques peuvent être associées aux anomalies morphologiques :
- l’ostéoporose, le plus souvent asymptomatique (ostéodensitométrie) mais parfois responsable de fractures pathologiques, volontiers costales ou vertébrales (cf. Item 128 – Ostéopathies fragilisantes) ;
- des troubles gonadiques par déficit gonadotrope :
- spanioménorrhée, voire aménorrhée secondaire, sans bouffées de chaleur chez la femme ;
- baisse de la libido et impuissance chez l’homme ;
- l’hypertension artérielle, généralement modérée ;
- des troubles psychiatriques de nature variable :
- irritabilité ;
- anxiété ;
- insomnie nocturne ;
- tendance dépressive ;
- exceptionnellement, tableau psychiatrique aigu, à type de psychose hallucinatoire et tendance suicidaire, qui se voit surtout lors des hypercortisolismes intenses.
3 Anomalies biologiques non spécifiques
L’intolérance aux glucides est fréquente ; le diabète sucré est retrouvé chez un tiers des patients lorsque l’on fait une HGPO.
4 Diagnostic biologique
Le diagnostic biologique permet d’affirmer l’hypercortisolisme.
Mise en évidence de la sécrétion excessive de cortisol
Le dosage plasmatique du cortisol matinal n’a pas d’intérêt diagnostique (chevauchement des valeurs normales et de celles rencontrées dans le syndrome de Cushing) : une cortisolémie normale le matin n’élimine pas le diagnostic (+++) et, à l’inverse, en conditions de stress, la cortisolémie est souvent élevée, pouvant faussement faire évoquer un syndrome de Cushing. De plus, la contraception œstroprogestative augmente artificiellement la cortisolémie source d’erreur diagnostique.
La mesure du cortisol libre urinaire (CLU) permet d’apprécier indirectement la quantité de cortisol produite sur l’ensemble du nycthémère (mesure réalisée sur plusieurs jours consécutifs car la sécrétion peut être fluctuante d’un jour à l’autre). Elle nécessite d’être bien expliquée au patient — jeter les premières urines de la miction du matin au lever le premier jour et commencer le recueil à partir de ce moment et ce jusqu’au lendemain matin en y intégrant les urines de la première miction du matin. Réalisée dans de bonnes conditions, elle est très pertinente. Malheureusement, c’est rarement le cas et elle peut s’avérer faussement normale ou faussement élevée.
Rupture du rythme circadien de sécrétion du cortisol
Le dosage du cortisol à minuit, moment où la concentration est physiologiquement minimale, ne peut être réalisé que dans le cadre d’une hospitalisation :
- une cortisolémie à minuit basse élimine un hypercortisolisme ;
- à l’inverse, une cortisolémie à minuit élevée est en faveur du diagnostic.
De plus en plus de centres proposent la réalisation de dosages du cortisol salivaire à minuit, qui a l’avantage de pouvoir être fait en ambulatoire mais n’est pas actuellement remboursé par l’assurance maladie.
Perte de rétrocontrôle
On observe la perte de rétrocontrôle des glucocorticoïdes exogènes sur la sécrétion d’ACTH hypophysaire (et donc de cortisol) suite à une absence de freinage.
Plusieurs modalités de « freinage » surrénalien peuvent être proposées. Elles utilisent un glucocorticoïde de synthèse très puissant, la dexaméthasone, qui n’est pas reconnue lorsque l’on dose le cortisol dans le sang ou dans les urines.
Test de freinage « minute » (+++)
Le test de freinage minute est le plus simple et peut être réalisé en ambulatoire.
La cortisolémie est mesurée le matin entre 6 et 8 h, après la prise orale de 1 mg de dexaméthasone la veille à 23 h.
Ce test permet de dépister la très grande majorité des syndromes de Cushing (grande sensibilité) mais sa spécificité est moins satisfaisante.
Des faux positifs (c’est-à-dire une absence de freinage) surviennent chez 10 à 20 % des sujets indemnes de syndrome de Cushing. On sera méfiant vis-à-vis de la contraception œstroprogestative qui augmente artificiellement la cortisolémie et des inducteurs enzymatiques qui accélèrent le métabolisme de la dexaméthasone.
Le test est en faveur du diagnostic d’hypercortisolisme non freinable si la cortisolémie ne s’abaisse pas au-dessous du seuil de 18 ng/ml [50 nmol/l].
Test de freinage « faible »
Ce test est également appelé freinage « standard ».
Une dose de 0,5 mg de dexaméthasone est administrée toutes les 6 heures (soit 2 mg par jour) pendant 2 jours. Les critères de jugement sont les mêmes que précédemment.
Stratégie d’exploration paraclinique
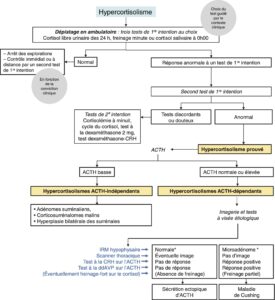
En dehors des cas cliniquement évidents, le diagnostic doit s’effectuer en deux phases (fig. 16) : tests de première intention puis tests de deuxième intention.
________________________________________________________________________________
Fig. 16
Schéma du diagnostic biologique et étiologique de l’hypercortisolisme. * En cas de doute (IRM normale et tests discordants) → Cathétérisme des sinus pétreux.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
En première intention, en cas de suspicion de syndrome de Cushing, un test doit être réalisé parmi les trois tests suivants : freinage minute par 1 mg de dexaméthasone, cortisol salivaire à minuit, cortisol libre urinaire des 24 heures.
On préfère : le cortisol libre urinaire des 24 heures en cas de syndrome de Cushing cliniquement évident voire sévère ; le freinage minute en cas de forme moins évidente en raison de sa très bonne sensibilité ; le cortisol libre urinaire des 24 heures ou le cortisol salivaire à minuit en cas de prise d’œstroprogestatifs.
Lorsqu’un test de première intention est normal, la réalisation immédiate ou à distance d’un second test de première intention sera discuté en fonction du degré de suspicion clinique.
Lorsqu’un test de première intention est anormal, la réalisation immédiate d’un second test de première intention est recommandée.
5 Diagnostic étiologique
Ce diagnostic permet d’affirmer l’origine hypophysaire de l’hypercortisolisme (cf. fig. 16).
L’hypercortisolisme étant établi (absence de freinage, absence de rythme nycthéméral du cortisol plasmatique et/ou CLU élevé), la première étape de l’enquête étiologique consiste à établir si l’hypercortisolisme dépend ou non de l’ACTH. Cette étape repose sur le dosage de l’ACTH plasmatique :
- une concentration d’ACTH effondrée est en effet évocatrice d’un syndrome de Cushing d’origine surrénalienne (adénome ou corticosurrénalome malin) ; elle impose alors la réalisation d’un scanner des surrénales ;
- des concentrations d’ACTHdans les valeurs normales (non effondrées, c’est-à-dire inappropriées en présence d’un hypercortisolisme) ou élevées sont en faveur d’un syndrome de Cushing ACTH-dépendant.
Si le syndrome de Cushing est ACTH-dépendant, il faut alors en affirmer l’origine hypophysaire (adénome corticotrope) et éliminer une sécrétion ectopique non hypophysaire d’ACTH.
Cette étape peut être particulièrement difficile car les adénomes corticotropes responsables de la maladie de Cushing sont généralement de petite taille (microadénomes). Leur mise en évidence à l’IRM peut donc ne pas être possible. Par ailleurs, les tumeurs neuroendocrines non hypophysaires responsables d’une sécrétion ectopique d’ACTH sont parfois elles aussi de très petite taille et difficiles à mettre en évidence avec les moyens radiologiques conventionnels (tumeurs carcinoïdes « occultes »). Dans la mesure où les concentrations d’ACTH sont souvent dans les mêmes zones au cours des deux pathologies, il faut souvent avoir recours à des tests plus sophistiqués. Les arguments en faveur de l’adénome hypophysaire corticotrope à l’origine de l’hypersécrétion d’ACTH (maladie de Cushing) sont :
- la présence d’un microadénome à l’IRM (dans la moitié des cas seulement), beaucoup plus rarement d’un macroadénome ;
- le résultat de différents tests biologiques, qui peuvent être utilisés pour distinguer les adénomes hypophysaires des tumeurs ectopiques ACTH-sécrétantes :
- test de freinage fort (2 mg de dexaméthasone toutes les 6 heures pendant 2 jours ou 8 mg de dexaméthasone en une prise à minuit) ;
- test à la CRH ;
- test à la dDAVP (desmopressine) ;
- en cas de doute, un cathétérisme des sinus pétreux inférieurs avec dosage de l’ACTH peut être réalisé.
6 Éliminer les « pseudo-Cushing » par hypercortisolisme fonctionnel
Le principal diagnostic différentiel des syndromes de Cushing ACTH-dépendants est constitué par les « pseudo-Cushing » par hypercortisolisme fonctionnel ACTH-dépendant modéré.
Ce sont le stress intense, les dépressions sévères, les psychoses et l’alcoolisme qui activent l’axe corticotrope et qui s’accompagnent d’une résistance relative et réversible aux glucocorticoïdes.
Ces « pseudo-Cushing » sont marqués cliniquement par un syndrome de Cushing modéré, avec des signes cliniques cataboliques (cutanés, musculaires, osseux) faibles ou absents. Biologiquement, les patients présentent :
- des anomalies modérées aux tests de première ligne (cortisol libre urinaire, freinage minute par 1 mg de dexaméthasone) ;
- une concentration plasmatique d’ACTH qui n’est pas freinée.
L’épreuve du temps et la réévaluation clinique et biologique des patients à distance, éventuellement après mise en route d’un traitement psychotrope adapté ou après sevrage alcoolique, permettent souvent de trancher.
IV Découverte de l’adénome hypophysaire devant un tableau d’insuffisance antéhypophysaire
La présence d’un adénome hypophysaire peut être à l’origine d’une insuffisance antéhypophysaire. Le diagnostic est rendu difficile par le fait que son début est souvent insidieux.
________________________________________________________________________________
Attention !
Un adénome hypophysaire ne s’accompagne jamais d’un diabète insipide (sauf en postopératoire ou à l’occasion d’une apoplexie hypophysaire). La présence d’un diabète insipide chez un patient porteur d’une lésion hypothalamo-hypophysaire doit donc impérativement faire rechercher une autre étiologie que l’adénome hypophysaire (cf. supra le diagnostic différentiel des adénomes en imagerie) (+++).
________________________________________________________________________________
A Aspect clinique caractéristique du panhypopituitarisme chez l’adulte
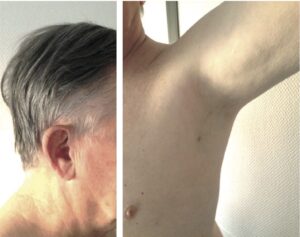
Le faciès est pâle, un peu « vieillot ». La dépigmentation des aréoles mammaires et des organes génitaux externes est constante. La peau est mince, froide, sèche. Les rides au niveau du front et de la partie externe des orbites sont fines. Les cheveux sont fins et soyeux. La dépilation est complète au niveau des aisselles et du pubis (signe intéressant chez les sujets de moins de 60 ans), consécutive à l’absence d’androgènes surrénaliens et gonadiques (fig. 17).
________________________________________________________________________________
Fig. 17
Insuffisance antéhypophysaire marquée par l’absence de barbe, des cheveux fins et secs, des rides fines, une dépilation axillaire et pubienne.
(Source : CEEDMM, 2021.)
________________________________________________________________________________
B Signes liés aux déficits des fonctions hypophysaires
1 Signes liés à l’insuffisance gonadotrope
Chez l’homme
À l’interrogatoire, on note une disparition de la libido, responsable parfois de troubles de l’érection. À l’examen, la pilosité du visage est raréfiée, les testicules sont petits et mous à la palpation. Le patient est généralement infertile.
Chez la femme
L’aménorrhée, précédée parfois d’irrégularités menstruelles classiquement sans bouffées de chaleur, est un signe extrêmement fréquent. La patiente est généralement infertile. La carence en œstradiol est à l’origine d’une atrophie des muqueuses vaginales et vulvaires, responsable d’une dyspareunie.
Dans les deux sexes
L’hypogonadisme prolongé provoquera une déminéralisation osseuse et une ostéoporose.
Autres signes
Lorsque le déficit est apparu avant la puberté, au tableau clinique précédent s’associe la présence d’un impubérisme (absence de puberté) ou d’un retard pubertaire, voire d’un retard de croissance si le déficit est associé à un déficit en GH.
2 Signes liés au déficit corticotrope
Ce déficit est responsable d’une asthénie importante et d’une tendance à l’hypotension. Un amaigrissement est souvent noté, associé à une anorexie. Il expose à un risque d’hypoglycémie de jeûne et d’hyponatrémie.
Le déficit en cortisol est responsable d’une réduction de la néoglucogenèse hépatique et donc d’un risque d’hypoglycémie de jeûne.
Inversement, la couche glomérulée de la surrénale, qui est sous le contrôle de l’axe rénine-angiotensine, n’est pas affectée par le déficit en ACTH ; il n’y a, par conséquent, pas de déficit sévère en aldostérone. Il n’y a donc pas de perte de sel ni de tendance à l’hyperkaliémie et à l’acidose lors des déficits corticotropes, contrairement aux insuffisances surrénaliennes primitives (cf. Item 245 – Insuffisance surrénale).
De même, l’hyponatrémie observée dans les insuffisances hypophysaires est une hyponatrémie de dilution liée à une sécrétion inappropriée d’ADH, non associée à un déficit volémique et donc sans insuffisance rénale fonctionnelle, contrairement à ce qui est observé dans la maladie d’Addison (cf. Item 267 – Désordres hydroélectrolytiques). Le déficit corticotrope est parfois très bien toléré et celui-ci n’est alors découvert que lors de l’évaluation hormonale d’un patient porteur d’une lésion hypothalamo-hypophysaire. Cependant, le déficit corticotrope peut mettre la vie en danger par un collapsus cardiovasculaire vasoplégique dans la mesure où le patient perd sa réponse vasopressive au stress — les facteurs de décompensation sont en particulier infectieux, traumatique ou chirurgical —, qu’il développe des hypoglycémies et qu’une hyponatrémie peut apparaître.
3 Signes liés au déficit thyréotrope
Ce déficit entraîne une carence en hormones thyroïdiennes d’intensité variable, mais très souvent moins sévère que celle observée au cours des hypothyroïdies périphériques. Les signes d’hypothyroïdie sont par conséquent d’intensité modérée.
4 Signes liés au déficit somatotrope
Chez l’adulte
L’absence de sécrétion de GH n’a pas de conséquence clinique évidente, sauf une diminution de la masse et de la force musculaires, une tendance à l’adiposité abdominale, une fatigue et une diminution de la qualité de vie.
Chez l’enfant (+++)
Le déficit en GH est responsable d’un retard de croissance (pour le détail des signes cliniques et biologiques : cf. Item 53 dans le référentiel de Pédiatrie du Collège national des professeurs de pédiatrie). Les accidents hypoglycémiques chez l’enfant, conséquence des déficits somatotrope et corticotrope, sont très fréquents et souvent révélateurs.
C Bilan hypophysaire fonctionnel
Ce bilan permet de confirmer le diagnostic clinique d’insuffisance antéhypophysaire.
1 Déficit corticotrope
Le test de référence pour la mise en évidence d’un déficit corticotrope est l’hypoglycémie insulinique.
Hypoglycémie insulinique
En cas d’insuffisance corticotrope, le cortisol ne s’élève pas au-delà de 185 ng/ml [500 nmol/l], à condition que la glycémie au cours de l’hypoglycémie insulinique se soit abaissée à moins de 2,2 mmol/l [0,40 g/l] — mais une hypoglycémie est parfois difficile à obtenir chez l’obèse. Elle est contre-indiquée en cas d’insuffisance coronarienne et de comitialité.
Autres tests
Compte tenu des inconvénients de l’hypoglycémie insulinique, d’autres tests plus simples sont parfois utilisés.
Simple dosage de la cortisolémie
Les performances de ce dosage seul sont médiocres, ce qui explique qu’un test dynamique soit indispensable dans la majorité des cas. Néanmoins, une cortisolémie du matin inférieur à 50 ng/ml [140 nmol/l] affirme l’insuffisance corticotrope et une cortisolémie au-dessus de 135 ng/ml [365 nmol/l] l’élimine.
Entre les deux, seuls des tests dynamiques permettront d’affirmer ou d’infirmer le diagnostic d’insuffisance corticotrope.
Test au Synacthène® immédiat 0,250 mg
Le seul critère de réponse normale (affirmant l’intégrité corticotrope) est un seuil de cortisolémie après Synacthène® immédiat > 180 ng/ml [500 nmol/l] — le seuil est adapté à la méthode de dosage.
Son rationnel repose sur l’atrophie corticosurrénale qui s’installe en cas de déficit prolongé (> 3 mois) : dans ce cas, la surrénale qui est saine répondra à une injection ponctuelle d’ACTH mais de manière insuffisante (< 180 ng/ml). Dans la période postopératoire immédiate ou chez les patients ayant une insuffisance corticotrope partielle, la réponse peut être faussement normale.
2 Déficit thyréotrope
Le déficit en TSH ne peut pas être mis en évidence par un dosage de TSH — concentrations de TSH le plus souvent normales chez les patients présentant une authentique hypothyroïdie secondaire ou déficit thyréotrope.
Le seul dosage permettant réellement de faire le diagnostic d’hypothyroïdie d’origine hypothalamo-hypophysaire est donc la mise en évidence d’une diminution de la concentration plasmatique de T4 libre, sans élévation de celle de TSH. La mesure de la T3 libre est moins utile car elle est fréquemment normale.
3 Déficit gonadotrope
Chez la femme
Avant la ménopause
Le diagnostic d’une insuffisance gonadotrope est essentiellement clinique : il est établi sur l’existence d’une aménorrhée ou d’une oligoménorrhée associées à des signes de déprivation œstrogénique (baisse de la libido, sécheresse vaginale, dyspareunie, etc.). Typiquement, l’œstradiol plasmatique est bas, alors que les gonadotrophines, en particulier la FSH, ne sont pas élevées (parfois basses ou dans les valeurs « normales »).
Les tests dynamiques (test à la GnRH, anciennement dénommée LHRH) n’ont pas d’intérêt.
Après la ménopause
Le diagnostic est établi sur le dosage basal des gonadotrophines : elles sont basses ou dans les valeurs correspondant aux femmes jeunes, alors qu’on les attend élevées chez la femme ménopausée.
Chez l’homme
Le diagnostic d’hypogonadisme hypogonadotrophique est établi sur la présence de troubles sexuels (baisse de la libido) associés à une concentration basse de testostérone, sans élévation des gonadotrophines (en particulier de FSH) qui sont basses ou dans les valeurs « normales ».
Le test à la GnRH n’offre aucun intérêt diagnostique.
Il faut aussi savoir que l’hyperprolactinémie peut, en soi, être responsable d’un déficit gonadotrope fonctionnel (cf. supra). Dans ce cas, la correction de l’hyperprolactinémie permet de restaurer une fonction gonadotrope et donc gonadique normale.
4 Déficit somatotrope
Déficit en hormone de croissance chez l’enfant (+++)
Le diagnostic est établi devant un retard de croissance et une absence de réponse adéquate à la stimulation de la GH par différents tests, en particulier celui de l’hypoglycémie insulinique.
Chez l’adulte
Le déficit en GH est le plus fréquent de tous les déficits hypophysaires puisqu’il est présent dès qu’une, au moins, des autres hormones antéhypophysaires est déficiente. Faire le diagnostic de déficit en hormone de croissance n’a réellement d’intérêt que dans l’hypothèse de la mise en route d’un traitement par GH chez l’adulte. Si un traitement par GH est envisagé, il faut pouvoir disposer des résultats d’au moins deux tests de stimulation de la GH. Les tests généralement recommandés chez l’adulte sont l’hypoglycémie insulinique ou le test associant la GHRH (Growth Hormone Releasing Hormone) avec l’arginine.
D Diabète insipide
Le diabète insipide central peut survenir après chirurgie d’un adénome hypophysaire ou bien en cas de lésion non adénomateuse de la région hypothalamo-hypophysaire. Il est caractérisé par une polyurie hypotonique supérieure à 3 litres par 24 heures chez l’adulte. Sa présence avant une chirurgie hypophysaire élimine le diagnostic d’adénome hypophysaire et fait plutôt évoquer les diagnostics différentiels évoqués plus haut.
Le diabète insipide est :
- le plus souvent secondaire à un défaut de synthèse de la vasopressine (ou ADH) : diabète insipide central ;
- parfois à une résistance à la vasopressine : diabète insipide néphrogénique ;
- une anomalie de la soif (polydipsie primaire) ;
- ou une destruction précoce de la vasopressine par une enzyme placentaire (diabète insipide gestationnel, très rare).
Un interrogatoire minutieux doit rechercher :
- la persistance nocturne de la polyurie, bon signe d’organicité ;
- préciser le début et l’ancienneté des troubles ;
- la présence de réveils nocturnes ;
- la notion de prise médicamenteuse ;
- le caractère éventuellement familial du trouble.
L’examen clinique nécessite la mesure du poids, de la pression artérielle et, surtout, la quantification nycthémérale des boissons et de la diurèse. Des signes de déshydratation, un éventuel globe vésical, des signes d’hypersécrétion ou d’hyposécrétion hormonale hypophysaire, un syndrome tumoral, des signes de granulomatose ou de cancer doivent être recherchés.
Dans les formes sévères, le diagnostic de diabète insipide est porté sur la clinique et des dosages biologiques de base (osmolalité urinaire < 200 mOsmol/kg H2O et natrémie > 145 mmol/l), le plus souvent associé à un test thérapeutique à la desmopressine et à une IRM hypophysaire.
Dans les formes partielles (osmolalité urinaire comprise entre 300 et 800 mOsmol/kg H2O), le test de restriction hydrique garde un intérêt, couplé au dosage de la vasopressine ou de la copeptine. Ces examens doivent être faits en milieu spécialisé.
Le diabète insipide central par déficit en vasopressine doit être différencié des diabètes insipides néphrogéniques par résistance rénale à la vasopressine (syndrome polyuro-polydipsique à vasopressine élevé). Les diabètes insipides néphrogéniques sont parfois familiaux et génétiquement déterminés mais le souvent plus acquis et secondaires à :
- une affection rénale ;
- une hypercalcémie ;
- une hypokaliémie ;
- une cause iatrogène : lithium (+++), 12 à 40 % des cas.
L’IRM hypophysaire recherche d’une part un hypersignal spontané de la posthypophyse signant la présence de vasopressine, d’autre part une anomalie de l’hypophyse ou de la tige hypophysaire.
Un diabète insipide central acquis de révélation brutale doit évoquer un craniopharyngiome ou un germinome avant 30 ans et une métastase après 50 ans. Les traumatismes crâniens se compliquent dans 15 à 20 % des cas d’hypopituitarisme, dont 2 % de diabète insipide.
Les principales causes de diabète insipide sont résumées dans le tableau 2.
________________________________________________________________________________
Tableau 2
Causes des diabètes insipides centraux par déficit en vasopressine. (Source : CEEDMM, 2021.)
| Acquis | Tumorale :
• < 30 ans : craniopharyngiome, germinome • > 50 ans : métastase (++) Post-traumatique ou post-chirurgical Inflammatoire, auto-immune, granulomateuse (histiocytose X chez l’enfant ; sarcoïdose chez l’adulte) Infectieuse Ischémique ou anoxique : choc, syndrome de Sheehan Syndromes malformatifs ou dégénératifs Toxique Idiopathique |
| Familiaux | Génétiquement déterminés, très rares |
________________________________________________________________________________
Le traitement des diabètes insipides centraux repose sur l’apport d’un substitut de la vasopressine, la desmopressine. En son absence, il est impératif que le patient boive autant qu’il le souhaite (souvent plusieurs litres par jour afin d’éviter une déshydratation sévère caractérisée biologiquement par une hypernatrémie).
E Imagerie
La mise en évidence d’un déficit hypophysaire isolé ou multiple impose la réalisation d’une IRM à la recherche d’une lésion hypothalamo-hypophysaire.
________________________________________________________________________________
Points-clés
- Un adénome hypophysaire avec expansion suprasellaire peut comprimer le chiasma optique et donner des troubles visuels à type d’hémianopsie bitemporale.
- L’IRM est l’examen de référence et met en évidence soit un microadénome (< 10 mm de diamètre) soit un macroadénome (> 10 mm).
- L’hyperprolactinémie est souvent secondaire à un adénome à prolactine, mais elle peut aussi être consécutive à la prise de médicaments (+++) ou à la présence d’une tumeur non prolactinique comprimant la tige pituitaire (hyperprolactinémie de déconnexion).
- Le diagnostic d’acromégalie, suspecté cliniquement, repose sur la mise en évidence d’une augmentation de la concentration d’IGF-1 et, éventuellement, sur l’absence de freinage de la GH à moins de 0,4 ng/ml après HGPO.
- La découverte d’un hypercortisolisme (syndrome de Cushing) est prouvée par au moins deux anomalies biologiques telles que l’augmentation du cortisol libre urinaire, la présence d’un cortisol salivaire vespéral élevé et/ou le freinage à la dexaméthasone (freinage minute) anormal. Elle impose un diagnostic étiologique reposant sur l’ACTH : si l’ACTH est élevée ou normale (non effondrée), il peut s’agir d’un adénome hypophysaire à ACTH (maladie de Cushing) ou d’une sécrétion ectopique d’ACTH.
- Une insuffisance antéhypophysaire doit être recherchée chez tout patient porteur d’un adénome hypophysaire. Sur le plan hormonal, le diagnostic repose sur une absence de réponse satisfaisante du cortisol et de la GH aux tests de stimulation, et sur une baisse de la T4 libre et des stéroïdes sexuels (testostérone chez l’homme, œstradiol chez la femme), respectivement sans élévation de la TSH ou des gonadotrophines.
- Un adénome hypophysaire ne s’accompagne jamais d’un diabète insipide (sauf en postopératoire ou à l’occasion d’une apoplexie hypophysaire). La présence d’un diabète insipide chez un patient porteur d’une lésion hypothalamo-hypophysaire doit donc impérativement faire rechercher une autre étiologie que l’adénome hypophysaire (+++).
________________________________________________________________________________
© CEEDMM – Août 2022.