|
Traitement médical des adénomes à prolactine : faut-il craindre un « effet Mediator® » ?
Gérald Raverot (Fédération d’endocrinologie du Pôle Est, groupement hospitalier Est, Lyon. gerald.raverot@chu-lyon.fr)
Au cours de l’été (juillet 2010), sans concertation avec les endocrinologues, sont apparues de nouvelles recommandations concernant la prescription de la cabergoline (1). En effet, il est maintenant demandé de faire des échographies cardiaques avant toute mise en route d’un traitement par cabergoline, à renouveler 3 à 6 mois après l’initiation du traitement puis tous les 6 à 12 mois, et ce indépendamment de la dose prescrite et de l’indication. S’agit-il d’une mesure parapluie en réaction aux remous autour du benfluorex ?
S’il semble légitime d’alerter le monde médical sur le risque potentiel de la cabergoline, et à un moindre degré de la bromocriptine, n’oublions pas que nos objectifs thérapeutiques ne sont pas de lutter contre quelques kilos excessifs mais bien contre une tumeur responsable d’infertilité, d’hypogonadisme, voire de troubles visuels chez des patients n’ayant souvent pas d’autre alternative thérapeutique. En effet, comme l’indiquent les recommandations de la Société française d’endocrinologie (2), de la Pituitary Society (3) et de l’Endocrine Society (4), le traitement de première intention des adénomes à prolactine est le plus souvent un traitement médical qui repose sur l’utilisation d’agonistes dopaminergiques. Or, ces agonistes dopaminergiques sont pour l’essentiel des dérivés de l’ergot de seigle et possèdent une activité sérotoninergique 5HT2B potentiellement responsable de valvulopathies cardiaques. Ainsi, en 2007, une publication du New England Journal of Medicine (5) a mis en évidence une corrélation entre la dose cumulée de cabergoline et la survenue de valvulopathies cardiaques chez des sujets parkinsoniens, avec toutefois des doses cumulées moyennes très élevées (2341 ± 2039 mg pour les valvulopathies de grade 0 à 2 et 4015 ± 3208 mg pour les grades 3 et 4). À la suite de cette publication, de nombreuses études aux méthodologies très diverses ont abouti à des résultats discordants. Bien qu’il soit moindre, le risque semble aussi exister pour la bromocriptine (6).
Une revue récente de ces études, publiée dans le Journal of clinical endocrinology and metabolism (7), démontre qu’il n’y a pas de risque accru de régurgitation valvulaire associé à la cabergoline. Toutefois, une étude conclut à un risque accru de régurgitation tricuspide d’intensité légère à modérée (8). En outre, les résultats de cette revue suggèrent une corrélation entre la survenue de régurgitation et la dose cumulée moyenne de cabergoline. Ainsi, bien qu’il n’y ait pas d’association évidente entre l’utilisation des agonistes de la dopamine et la survenue de valvulopathies cardiaques dans cette population, les auteurs suggèrent qu’une attention particulière soit portée aux patients nécessitant un traitement prolongé avec de fortes doses de cabergoline, sans pour autant préciser de seuil. En outre, M.E. Molitch proposait qu’une surveillance soit envisagée pour les patients traités au long cours avec une dose supérieure à 2 mg/semaine (9). Les conclusions de cette revue sont de conseiller l’utilisation de doses de cabergoline les plus faibles possible et de proposer une surveillance échocardiographique uniquement au cas par cas, et non pas de manière généralisée. Ces conclusions viennent d’être renforcées par celles du consensus de l’Endocrine Society (4) qui précise que « une augmentation des doses doit être progressive et guidée par des taux de prolactine. Chez les patients qui ont besoin de très fortes doses pendant de longues périodes, l’échocardiographie peut être nécessaire pour évaluer les anomalies valvulaires. Bien que la dose et la durée précise ne peuvent être identifiées à ce moment, les patients recevant des doses habituelles de cabergoline (1-2 mg/semaine) n’ont probablement pas besoin de dépistage échographique systématique« . Il ne faut pas non plus oublier que la population générale présente également une « prévalence élevée » de survenue de valvulopathies, ce qui est bien rappelé dans cette revue très récente que Philippe Chanson met à notre disposition en avant-première (10).
Ces conclusions font donc appel au bon sens clinique et à la prudence mesurée. En l’absence de preuve formelle de dangerosité ou d’innocuité dans cette indication, elles ont le mérite de souligner le risque sans toutefois le surévaluer. Elles placent la surveillance échographique de ces patients dans une prise en charge individuelle et adaptée au patient (âge, dose et durée prévisible de traitement), au contraire du nouveau RCP de la cabergoline qui ne tient pas compte de la personnalisation de la médecine ni de l’évaluation du rapport coût/bénéfice d’une telle surveillance généralisée. S’il est raisonnable de proposer un contrôle échographique cardiaque systématique en début de traitement par cabergoline, la fréquence ultérieure des contrôles doit être individualisée en fonction du contexte clinique.
Références bibliographiques
1. http://afssaps-prd.afssaps.fr/php/ecodex/frames.php?specid=69671845&typedoc=R&ref=R0158677.htm
2. Brue T, Delemer B, Bertherat J et al. Diagnostic et prise en charge des hyperprolactinémies. Consensus d’experts de la Société Française d’Endocrinologie (SFE). Médecine Clinique endocrinologie & diabète 2006;Hors-série:1-7. http://www.sfendocrino.org/_images/mediatheque/articles/pdf/Brue2006MCEDconsensusHPRL.pdf
3. Casanueva FF, Molitch ME, Schlechte JA et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol 2006;65(2):265-73.
4. Melmed S, Casanueva FF, Hoffman AR et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011;96(2):273-88.
5. Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med 2007;356(1):39-46.
6. Sherlock M, Toogood AA, Steeds R. Dopamine agonist therapy for hyperprolactinaemia and cardiac valve dysfunction; a lot done but much more to do. Heart 2009;95(7):522-3.
7. Valassi E, Klibanski A, Biller BM. Clinical Review#: Potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab 2010;95(3):1025-33.
8. Colao A, Galderisi M, Di Sarno A et al. Increased prevalence of tricuspid regurgitation in patients with prolactinomas chronically treated with cabergoline. J Clin Endocrinol Metab 2008;93:3777-84.
9. Molitch ME. The cabergoline-resistant prolactinoma patient: new challenges. J Clin Endocrinol Metab 2008;93(12):4643-5.
10. Maione L, Hescot S, Young J, Chanson P. Agonistes dopaminergiques et valvulopathies : quels risques ? Médecine Clinique endocrinologie & diabète 2011;51:1-5.
|
|
Anticorps anti-PIT-1 et déficit hypophysaire multiple de l’adulte
Frédéric Castinetti (Service d’endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Yamamoto M, Iguchi G, Takeno R et al. Adult combined GH, prolactin, and TSH deficiency associated with circulating PIT-1 antibody in humans. J Clin Invest 2011;121(1):113-9.
PIT-1 (POU1F1) est un facteur de transcription impliqué dans les étapes terminales de différentiation hypophysaire qui aboutissent aux cellules somatolactotropes (GH) et thyréotropes (TSH). Plusieurs mutations de POU1F1 ont été rapportées chez l’homme comme étant responsables de déficit hypophysaire congénital en GH, en prolactine et en TSH. Une expression de PIT-1 est retrouvée dans l’hypophyse à l’âge adulte sans que son rôle soit précisément déterminé.
L’équipe de M. Yamamoto rapporte le cas de 3 patients âgés de 44, 75 et 78 ans qui présentent un déficit somatolactotrope et thyréotrope associé, dans 1 cas, à une hypoplasie hypophysaire et, dans les 2 autres cas, à une IRM normale. Deux de ces patients présentaient un terrain auto-immun sous-jacent avec, pour 1 patient, une gastrite atrophique (associée à une probable insuffisance surrénalienne auto-immune) et, pour l’autre patient, un diabète de type 1. L’interrogatoire ne retrouvait aucun argument en faveur d’un déficit hypophysaire congénital. Une recherche de mutation de PIT-1, PROP1 et HESX1 s’est révélée négative. Dans le sérum des patients, les auteurs ont mis en évidence un anticorps IgG spécifique reconnaissant le domaine de transactivation de PIT-1. Cet anticorps n’a pas été retrouvé dans le sérum de patients témoins porteurs de diverses maladies auto-immunes, incluant des hypophysites. Une autopsie a été réalisée sur 1 des 3 patients décédés : l’analyse en immunohistochimie de coupes hypophysaires n’a pas permis de mettre en évidence de marquage dirigé contre les cellules somatotropes, lactotropes et thyréotropes. L’hypophyse était fibreuse, avec une infiltration lymphocytaire. Cette infiltration lymphocytaire était également retrouvée au sein de la majorité des autres organes endocrines. Compte tenu de l’association à d’autres pathologies auto-immunes, les auteurs suggèrent que ces patients pourraient présenter une polyendocrinopathie auto-immune dont le type reste à définir.
En conclusion, l’hypothèse d’anticorps anti-PIT-1 est intéressante. Le seul écueil est qu’il n’a jamais été démontré que PIT-1 exerçait le même rôle au cours de l’embryogenèse et dans l’hypophyse adulte. En d’autres termes, PIT?1 permet-il la différentiation de cellules souches hypophysaires potentielles à l’âge adulte chez l’homme ? De plus, les auteurs ont décrit une hypophyse inflammatoire (hypophysite ?) et des anticorps anti-PIT-1, mais ils n’ont pas la preuve formelle de l’implication de ces derniers dans la pathologie. Les modèles murins permettraient éventuellement d’apporter un argument supplémentaire : l’injection d’anticorps anti-PIT-1 chez une souris adulte sans déficit hypophysaire aboutit-elle à un phénotype hypopituitaire ?
……………………………………………………………………………………………………………………………………………………………
Accident vasculaire cérébral et hypopituitarisme
Frédéric Castinetti (Service d’endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Bondanelli M, Ambrosio MR, Carli A et al. Predictors of pituitary dysfunction in patients surviving ischemic stroke.
J Clin Endocrinol Metab 2010;95(10):4660-8.
Existe-t-il un risque d’hypopituitarisme chez les patients ayant présenté un accident vasculaire cérébral (AVC) ischémique ? L’équipe de M. Bondanelli a mené une étude prospective sur 56 patients suivis dans le centre de Ferrara, en Italie. La cohorte était composée de 34 hommes et de 22 femmes, de 64,8 ans d’âge moyen. L’échelle de classification NIHSS (National Institute of Health Stroke Score) a permis de classer les patients en 2 groupes, selon la sévérité du déficit. Un bilan endocrinologique (effectué à 8 heures du matin avec IGF-1 et test couplé GHRH-arginine, TSH/T4, ACTH/cortisol et test au synacthène 1 µg, LH-FSH/testostérone/œstradiol et prolactine) était effectué 1 à 3 mois après l’AVC, puis répété 12 à 15 mois après. Seuls 4 patients ont été perdus de vue entre ces 2 visites de suivi.
Les résultats affichés par les auteurs sont :
– à la première visite : 17 patients avec un « déficit » isolé en GH (réponse insuffisante au test mais taux d’IGF-1 strictement normal chez 15 patients), 6 avec un déficit gonadotrope isolé et 1 avec un déficit corticotrope. Trois de ces patients présentaient un déficit multiple (ACTH, GH, LH/FSH dans 1 cas ; GH et LH/FSH dans 2 cas) ;
– à la deuxième visite : « déficit » en GH persistant chez 14 patients (taux d’IGF-1 strictement normal chez 11 patients), déficit gonadotrope persistant chez 3 patients et déficit corticotrope persistant chez le patient atteint à la première visite. Deux patients présentaient des déficits multiples.
Les auteurs ont ensuite recherché les facteurs prédictifs d’hypopituitarisme chez les patients ayant présenté un AVC : sévérité de l’AVC, atteinte cérébrale antérieure, présence d’une hémorragie sur le TDM de contrôle à 48 heures, terrain diabétique sous-jacent. Les auteurs affirment également que la présence d’un hypopituitarisme est corrélée à un plus mauvais pronostic fonctionnel, mais ce résultat est probablement biaisé par le fait que ces patients présentent aussi des atteintes ischémiques plus sévères. Que déduire de ces résultats ? En termes de déficit en GH, l’étude présente quelques incohérences, comme la dissociation IGF-1 normale et la réponse insuffisante à un test couplé (norme non définie dans le texte). De plus, la plupart de ces « déficits » en GH sont isolés et il est vraisemblable que les normes des tests de stimulation définies chez des sujets normaux ne soient pas nécessairement les mêmes que chez des sujets ayant présenté un AVC. Le diagnostic même de déficit en GH semble donc discutable dans la majorité des cas. Même si ce diagnostic était confirmé chez certains patients, la nécessité d’un traitement substitutif par GH semble plus que discutable. De même, le déficit gonadotrope n’est pas bien défini : chez des sujets âgés en moyenne de plus de 65 ans, quelles sont les normes de la testostérone utilisées ? et quelles valeurs basales de la LH et de la FSH ont été utilisées ? Il est donc vraisemblable que les chiffres avancés par les auteurs (environ 36 % d’hypopituitarisme chez les patients ayant présenté un AVC) sont largement surestimés. Reste les cas plus intéressants, mais finalement rares, des déficits multiples.
En conclusion, il est difficile, à partir d’une seule étude, de tirer des conclusions sur le risque d’hypopituitarisme après AVC. Il faudra des études menées avec une méthodologie plus stricte et des normes définies à partir de cette sous-population de patients ayant subi un AVC pour avoir une réponse définitive. En pratique et à ce jour, un dépistage systématique d’hypopituitarisme chez ces patients ne se justifie pas.
……………………………………………………………………………………………………………………………………………………………
Hypertension intracrânienne idiopathique, maladie de Cushing et déficit corticotrope
Frédéric Castinetti (Service d’endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Zada G, Tirosh A, Kaiser UB, Laws ER, Woodmansee WW. Cushing’s disease and idiopathic intracranial hypertension: case report and review of underlying pathophysiological mechanisms. J Clin Endocrinol Metab 2010;95(11):4850-4.
L’hypertension intracrânienne (HTIC) idiopathique est en général observée chez des femmes jeunes, obèses et présentant une symptomatologie d’hypertension intracrânienne (céphalées, nausées) sans obstacle mécanique cérébral. La survenue d’une HTIC idiopathique a été décrite sporadiquement dans le cadre de déficit en glucocorticoïdes, sans qu’un mécanisme physiopathologique ne puisse être clairement mis en cause.
L’équipe de G. Zada rapporte le cas d’une patiente de 33 ans ayant présenté une HTIC idiopathique dans les suites d’une reprise de chirurgie transsphénoïdale de maladie de Cushing. En post-opératoire immédiat, la patiente présentait un déficit corticotrope en faveur d’une rémission. Un traitement substitutif par hydrocortisone a été instauré. Le traitement, à raison de 15 mg/jour, a été poursuivi au long cours. Onze mois après la chirurgie, la patiente a présenté des nausées et des vomissements qui ont entraîné une absence de prise d’hydrocortisone pendant 48 heures, suivis d’un tableau typique d’hypertension intracrânienne avec céphalées et troubles visuels. L’examen ophtalmologique retrouvait un œdème papillaire bilatéral. L’IRM a éliminé l’éventualité d’une apoplexie de résidu hypophysaire ; la ponction lombaire n’a pas retrouvé d’arguments en faveur d’une méningite. Un diagnostic d’hypertension intracrânienne idiopathique a donc été posé et un traitement par dexaméthasone et acétazolamide, puis par prednisone et ponctions lombaires évacuatrices a été prescrit.
À ce jour, 12 cas ont été rapportés dans la littérature. L’HTIC idiopathique survenait généralement 2 à 4 semaines après la chirurgie, chez des patients sous hydrocortisone (à dose substitutive) dans la plupart des cas. On relève cependant 2 patients chez qui le diagnostic a été posé alors qu’ils n’étaient plus sous traitement substitutif. La physiopathologie reste incomprise : elle pourrait être due au démasquage d’un œdème cérébral sous-jacent provoqué par l’arrêt du traitement ou à une connexion entre glucocorticoïdes et drainage veineux cérébral. Les glucocorticoïdes modifient également la production, le flux et l’absorption du liquide céphalo-rachidien. Enfin, une expression d’aquaporine a été retrouvée au sein des plexus choroïdes ; cette expression est augmentée en cas d’arrêt des glucocorticoïdes, ce qui favoriserait la survenue d’une hypertension intracrânienne. Le traitement peut associer de l’acétazolamide, des diurétiques de l’anse, une augmentation des glucocorticoïdes, voire une chirurgie de dérivation ou de décompression. Le pronostic est en général bon.
En conclusion, le clinicien doit garder à l’esprit le rare mais possible diagnostic d’hypertension intracrânienne chez les patients opérés de maladie de Cushing et qui consultent pour céphalées et altération visuelle, après avoir éliminé la récidive locale (et en particulier l’apoplexie). Même si la survenue d’hypertension intracrânienne en cours de sevrage n’a jamais été décrite, il semble néanmoins prudent d’évoquer ce diagnostic en cas de survenue d’un événement aigu lors de l’arrêt, même bref, du traitement substitutif.
|
|
La radiochirurgie Gamma Knife®
Frédéric Castinetti (Service d’endocrinologie, diabète et maladies métaboliques, hôpital de la Timone, Marseille)
Le Gamma Knife® est une technique radiochirurgicale qui délivre les rayonnements issus de sources de cobalt en une seule session, avec une précision stéréotaxique, et a pour but de diminuer l’activité hypersécrétoire et/ou le volume tumoral de la cible. Cette technique s’oppose, par son principe, à la radiothérapie conventionnelle où la dose est fractionnée. Des études récentes ont souligné l’efficacité à moyen et à long termes (suivi à 10 ans) de cette procédure, avec une rémission dans environ 50 % des cas pour les adénomes sécrétants (GH, ACTH ou prolactine). Point surprenant, le risque de récidive tardive de maladie de Cushing : environ 10 à 15 % des patients considérés comme « guéris » par la radiochirurgie 2 à 3 ans après la procédure présentent une récidive 6 à 8 ans après. Ce type spécifique d’hypersécrétion nécessite donc une surveillance rapprochée. La radiochirurgie permet également une stabilisation ou une diminution du volume tumoral dans plus de 90 % des cas, ce qui souligne son intérêt dans les adénomes non sécrétants, plus particulièrement dans les résidus postchirurgicaux qui augmentent progressivement de volume.
Les principaux facteurs qui doivent orienter la décision thérapeutique vers une radiochirurgie sont :
– un faible volume tumoral ;
– une très bonne définition IRM de la cible (qui augmente les chances de rémission tout en diminuant le risque d’effets secondaires) ;
– de faibles niveaux hypersécrétoires (il existe une décroissance progressive des taux hormonaux jusqu’à l’obtention d’un plateau) ;
– une distance suffisante du chiasma (pour éviter un risque de névrite optique radio-induite).
Le principal inconvénient de la radiochirurgie est le délai pour obtenir une efficacité maximale : 2 à 4 ans environ, au cours desquels un traitement anti-sécrétoire efficace devra être proposé. Il faudra alors pratiquer régulièrement des sevrages pour déterminer l’efficacité de la procédure radiochirurgicale et décider de la poursuite ou non du traitement antisécréteur.
Le principal effet secondaire est la survenue d’un ou de plusieurs déficits hypophysaires, dans environ un quart des cas, en moyenne 2 à 8 ans après la radiochirurgie. Cela impose donc une surveillance rapprochée annuelle du patient au cours des 10 années qui suivent la procédure. Contrairement à la radiothérapie conventionnelle, il n’a pas été décrit de tumeurs secondaires chez les patients traités par Gamma Knife® pour un adénome hypophysaire. Des études avec un suivi plus long sont cependant nécessaires car leur survenue est retardée jusqu’à 20 ans après la radiothérapie (et les études de suivi de la radiochirurgie Gamma Knife® ne dépassent pas en moyenne les 10 ans). Autres effets secondaires potentiels, mais très rares : la survenue de paralysie oculomotrice ou de sténose carotidienne.
En pratique, la radiochirurgie a sa place dans l’arsenal thérapeutique de l’acromégalie et de la maladie de Cushing, probablement moins dans celui des prolactinomes (du fait de l’efficacité majeure des dopaminergiques et de la chirurgie). Elle doit cependant être réservée aux cas où la chirurgie est contre-indiquée (et préférentiellement pour les tumeurs de faible volume peu sécrétantes) ou partiellement efficace, avec une résistance ou une intolérance aux traitements médicamenteux. La radiochirurgie ne doit pas être opposée à la radiothérapie : les indications sont complémentaires car les profils de patients à traiter par l’une ou l’autre de ces modalités sont différents.
……………………………………………………………………………………………………………………………………………………………
|








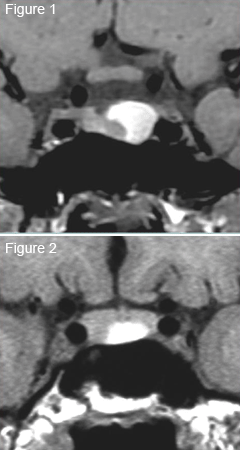 La mise en évidence en IRM d’une lésion intrasellaire hyperintense en T1 est souvent difficile à interpréter, et ce quelles que soient les données cliniques et biologiques.
La mise en évidence en IRM d’une lésion intrasellaire hyperintense en T1 est souvent difficile à interpréter, et ce quelles que soient les données cliniques et biologiques.