La lettre Surrénale de Juin 2016
 |
||||||||
| Cellules souches surrénaliennes, imagerie métabolique des phéos/PGL, diagnostic étiologique de l’hyperaldostéronisme primaire, hyperplasie congénitale : freiner l’ACTH sans glucocorticoïdes ? … | ||||||||
|
||||||||
|
Sous le signe de la transgression ! Chers passionnés de la surrénale, la Newsletter 7 vient bousculer vos certitudes. Médullo-cortico : ton voisin est ton frère, ennemi. L’imagerie se fait (de) la biochimie ! L’hypothalamus à la rescousse de l’hyperplasie congénitale : peut-on contrôler l’ACTH sans cortisol ? Et en image : Quand la surrénale fait mal. Une curieuse crise de colique néphrétique du 3e trimestre de la grossesse, qui nous vient de nos amis de Chambéry. Et pour finir, la page de pub : Sept articles et un article de synthèse en bonus, 47 recommandations, le tout dans le numéro du 13 juillet des Annales d’endocrinologie. Un véritable feu d’artifice ! |
||||||||
 |
||||||||
|
Quand la recherche sur les cellules souches surrénaliennes éclaircit la physiopathologie des néoplasies corticosurrénaliennes sécrétantes Les cellules souches sont retrouvées dans de nombreux tissus adultes où elles contribuent au renouvellement des cellules spécifiques des différents organes. L’équipe de S. Bornstein de l’Université de Dresde avait montré il y a 1 an, la présence de cellules souches multipotentes dans la médullosurrénale de souris capables de se différencier en neurones sympathiques ou en cellules chromaffines sous l’influence du stress (1). Ces cellules, qui partagent certaines caractéristiques phénotypiques avec les cellules gliales, sont identifiées par leur capacité à exprimer la nestine, un marqueur reconnu de cellules souches multipotentes, en particulier dans le système nerveux. Il est par ailleurs considéré que le renouvellement du cortex surrénalien s’effectue à partir de cellules progénitrices sous-capsulaires qui passent par différents stades de différenciation tout en migrant vers l’intérieur de la glande pour former successivement les zones glomérulée, fasciculée et réticulée. Lors de la 17th Adrenal Cortex Conference, qui s’est tenue à Boston du 29 au 31 mars dernier, la même équipe a montré que les cellules souches nestine-positives sont également présentes dans le cortex surrénalien murin, essentiellement dans la zone sous-capsulaire mais aussi de façon disséminée dans l’ensemble du cortex (2). À l’issue d’une immobilisation prolongée de plusieurs jours, ce qui représente un stress majeur pour la souris, ces cellules étaient capables d’exprimer la protéine Star (pour “steroidogenic acute regulatory protein”) traduisant leur faculté à se différencier également en cellules stéroïdogènes. L’ensemble de ces travaux laisse donc penser qu’il existe dans la glande surrénale des mammifères une population de cellules souches susceptibles de donner indifféremment naissance à des cellules corticales ou médullosurrénaliennes, en particulier sous l’influence du stress. Ces données, qui remettent en cause notre vision de l’origine et du renouvellement des deux tissus constitutifs de la glande surrénale, éclairent en outre d’un jour nouveau la physiopathologie de plusieurs types de lésions corticosurrénaliennes sécrétantes. Ainsi, l’expression anormale de marqueurs neuroendocrines notamment médullosurrénaliens par les cellules corticosurrénaliennes est bien documentée dans la dysplasie micronodulaire pigmentée des surrénales (DMPS), une cause rare de syndrome de Cushing (3). Cette affection est majoritairement due à des mutations de la sous-unité régulatrice alpha de la protéine kinase A (4). Dans la mesure où ces anomalies génétiques aboutissent à une activation constitutionnelle de la PKA mimant les effets d’un excès d’ACTH sur les cellules surrénaliennes tel qu’on peut l’observer au cours du stress, il est logique d’imaginer que les micronodules hyperplasiques de la DMPS pourraient correspondre à une expansion de cellules issues des cellules souches multipotentes décrites par S. Bornstein et son équipe. On peut aussi envisager que le phénotype à la fois stéroïdogène et neuroendocrine des cellules de DMPS puisse résulter de phénomènes de reprogrammation génétique touchant certaines cellules corticosurrénaliennes. De tels processus pourraient également intervenir dans la physiopathologie d’autres lésions néoplasiques corticosurrénaliennes comme l’adénome de Conn et l’hyperplasie macronodulaire bilatérale des surrénales au sein desquelles l’expression anormale de marqueurs neuroendocrines est bien établie (5, 6). À cet égard, il est intéressant de noter que la différenciation neuroendocrine illicite des cellules corticosurrénaliennes aboutit à la synthèse de neurotransmetteurs conventionnels et/ou de peptides bioactifs et de leurs récepteurs, constituant ainsi des boucles régulatrices autocrines/paracrines intra-surrénaliennes capables d’activer la stéroïdogenèse indépendamment de toute influence des facteurs régulateurs circulants, angiotensine II et ACTH, dont la sécrétion est d’ailleurs freinée par l’excès de corticostéroïdes (6). Ces systèmes stimulateurs intra-lésionnels constituent de ce fait des cibles intéressantes pour la mise au point de traitements pharmacologiques innovants des hypercorticismes pouvant représenter une alternative pertinente à la chirurgie surrénalienne. |
||||||||
|
Références bibliographiques 1. Rubin de Celis MF, Garcia-Martin R, Wittig Det al. Multipotent glia-like stem cells mediate stress adaptation. Stem Cells 2015;33(6):2037-51. 2. Rubin de Celis MF, et al. Is there a pan adrenal stem cell ? 17th Adrenal Cortex Conference, March 29-31, Boston, MA, USA. 3. Stratakis CA, Carney JA, Kirschner LS et al. Synaptophysin immunoreactivity in primary pigmented nodular adrenocortical disease: neuroendocrine properties of tumors associated with Carney complex. J Clin Endocrinol Metab 1999;84(3):1122-8. 4. Stratakis CA. New genes and/or molecular pathways associated with adrenal hyperplasias and related adrenocortical tumors. Mol Cell Endocrinol 2009;300(1-2):152-7. 5. Li Q, Johansson H, Kjellman M, Grimelius L. Neuroendocrine differentiation and nerves in human adrenal cortex and cortical lesions. APMIS 1998;106(8):807-17. 6. Lefebvre et al. Cell-to-cell communication in bilateral macronodular adrenal hyperplasia causing hypercortisolism. Front Endocrinol (Lausanne) 2015;6:34. |
||||||||
 |
||||||||
|
Vers l’imagerie métabolique des phéochromocytomes et des paragangliomes L’identification d’une mutation sur le gène SDHB, chez un patient avec paragangliome ou phéochromocytome, est un facteur de risque de malignité. Ce biomarqueur génétique diagnostique et pronostique n’est généralement pas disponible lors du diagnostic initial en raison des délais de rendu des tests génétiques. Dans la cellule tumorale, l’inactivation de la succinate déshydrogénase conduit à l’accumulation du succinate qui est un oncométabolite, et qui inhibe des enzymes importantes pour la régulation de l’angiogénèse et de la méthylation des histones et de l’ADN, processus impliqués dans la prolifération et l’agressivité tumorale. Il était jusqu’à présent possible de mesurer la concentration de succinate dans le tissu tumoral ou de réaliser un phénotypage immunohistochimique SDHx mais uniquement après que le patient a été opéré. Les avancées de C. Lussey-Lepoutre et al. ont été de montrer qu’il était possible de détecter l’accumulation de succinate dans la cellule tumorale in vivo par imagerie de résonance magnétique spectroscopique (1H-SRM), dans le cadre du bilan d’imagerie réalisé chez les patients atteints, avant la chirurgie (1). Cet examen, après qu’il a été validé dans une large cohorte prospective de patients, pourrait permettre de détecter les patients porteurs d’une mutation sur un gène SDHx ou de valider la fonctionnalité d’une variation génétique de signification inconnue mise en évidence lors de l’analyse moléculaire des gènes SDHx. L’ajout de cette séquence ne devrait pas trop majorer le temps de l’examen d’IRM notamment lors de l’exploration de la tête et du cou puisque G. Gravel et al. ont démontré, dans le cadre d’une étude ancillaire du PHRC PGL.EVA, qu’un temps d’examen raccourci de 40 à 10 minutes permettait de caractériser au mieux les lésions paraganglionnaires de la tête et du cou (2). Alors que les recommandations de suivi des patients atteints de phéochromocytomes et paragangliomes, récemment publiées, mettent l’accent sur le suivi à vie des patients porteurs d’une mutation sur le gène SDHB (3), il est vraisemblable que l’imagerie métabolique in vivo des phéochromocytomes et paragangliomes pourrait modifier à terme la prise en charge initiale de ces patients à risque. |
||||||||
|
Références bibliographiques 1. Lussey-Lepoutre C, Bellucci A, Morin A et al. In Vivo Detection of Succinate by Magnetic Resonance Spectroscopy as a Hallmark of SDHx Mutations in Paraganglioma. Clin Cancer Res 2016;22(5):1120-9. 2. Gravel G, Niccoli P, Rohmer V et al. The value of a rapid contrast-enhanced angio-MRI protocol in the detection of head and neck paragangliomas in SDHx mutations carriers: a retrospective study on behalf of the PGL.EVA investigators. Eur Radiol 2015 [Epub ahead of print] 3. Plouin PF, Amar L, Dekkers OM et al; Guideline Working Group. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016;174(5):G1-G10. |
||||||||
|
Diagnostic étiologique de l’hyperaldostéronisme primaire : le Pet scan aux inhibiteurs de CYP11B2 va-t-il remplacer le cathétérisme des veines surrénaliennes ? Dans la prise en charge de l’hyperaldostéronisme primaire, le diagnostic étiologique est une étape délicate : en effet, c’est elle qui doit distinguer les formes latéralisées d’hyperaldostéronisme primaire, qui peuvent bénéficier d’un traitement chirurgical, des formes bilatérales (ou “non latéralisées”) qui ne peuvent être traitées que médicalement. Actuellement la référence est le cathétérisme des veines surrénaliennes, en effet le scanner surrénalien manque de spécificité (sauf chez les rares patients de moins de 35 ans) et également de sensibilité, et la scintigraphie au noriodocholestérol est encore moins performante. Cependant le cathétérisme des veines surrénaliennes reste une exploration difficile, qui comprend des risques d’échecs (en particulier pour la veine surrénalienne droite) et des risques rares mais non nuls de complications : sa réalisation doit être réservée à des radiologues très entraînés. Une solution moins invasive et moins difficile serait donc la bienvenue et c’est peut-être l’avenir que dessinent T. Abe et al. dans un article très solide qui présente les résultats in vitro et chez l’animal d’une scintigraphie PET scan qui utilise un nouvel inhibiteur de l’adostérone synthase (CYP11B2), le CDP2230, marqué au Fluor 18 (1). Leur proposition n’est pas entièrement novatrice dans la mesure où des techniques similaires ont déjà été proposées avec un autre inhibiteur de CYP11B2, le métomidate, marqué au C11 (2), ou encore l’iodométomidate marqué à l’iode-123 (3). Le principe de ces scintigraphies est simple : elles utilisent un agent qui se lie à CYP11B2, enzyme exprimée spécifiquement dans les cellules qui synthétisent l’aldostérone. Le problème est que CYP11B2 est très proche de l’enzyme qui catalyse la dernière étape de synthèse du cortisol, la 11?-hydroxylase ou CYP11B1 et les inhibiteurs de CYP11B2 sont en général aussi capables d’inhiber CYP11B1. Or, dans l’hyperaldostéronisme primaire les tissus secrétant de l’aldostérone, que l’on cherche à identifier et qui expriment CYP11B2, sont entourés de tissu corticosurrénalien normal qui synthétise du cortisol et expriment CYP11B1 : il est donc capital de pouvoir s’affranchir d’une liaison avec CYP11B1. Pour cela les scintigraphies au métomidate ou iodométomidate nécessitent de freiner la sécrétion de cortisol, et l’expression de CYP11B1, par un prétraitement par dexaméthasone, ce qui rajoute une contrainte, et des effets indésirables potentiels, à l’exploration. L’interêt du nouvel agent utilisé par T. Abe et al. est qu’il se lie de façon beaucoup plus sélective à CYP11B2 que CYP11B1, ce qui permet d’espérer la réalisation de scintigraphie sans freinage. Nous n’en sommes pas encore là, mais dans cet article les auteurs montrent qu’in vitro, sur des coupes congelées d’adénomes de Conn humain étudiées par autoradiographie, le marquage par le 18F-CDP2230 montre un rapport signal/bruit de fond qui semble bien supérieur à celui du marquage 123 I-IMTO, et qui est bien corrélé au marquage immunohistochimique par anticorps anti-CYP11B2. In vivo les auteurs n’en sont qu’à l’animal, leur modèle étant le rat, qui exprime CYP11B1 et CYP11B2 dans sa corticosurrénale, mais les résultats semblent prometteurs : après injection de 18F-CDP2230, le rapport du signal entre la surrénale et un tissu avoisinant est bien plus élevé qu’après injection de 123 I-IMTO et le PET scan 18F-CDP2230 permet très bien de visualiser les surrénales du rat. Il reste cependant à démontrer qu’in vivo la fixation de 18F-CDP2230 permet bien de distinguer les tissus qui secrètent de l’aldostérone de ceux qui secrètent du cortisol car dans les images montrées ici , la définition du PET scan ne permet pas de distinguer si la fixation identifiée est spécifique de la fine couche glomérulée ou si elle concerne également la zone fasciculée. Il reste donc à espérer que des prochaines scintigraphies sur des modèles de tumeur de rat secrétant de l’aldostérone confirmeront les résultats in vitro, avant de passer à l’exploration des hyperaldostéronismes primaires chez l’homme. Cette technique devra alors être comparée au cathétérisme des veines surrénaliennes : l’avenir dira si les médecins nucléaires arriveront à reprendre le terrain du diagnostic étiologique de l’hyperaldostéronisme primaire, largement occupé par les radiologues interventionnistes. |
||||||||
|
Références bibliographiques 1. Abe T, Naruse M, Young WF Jr et al. A Novel CYP11B2-Specific Imaging Agent for Detection of Unilateral Subtypes of Primary Aldosteronism. J Clin Endocrinol Metab 2016;101(3):1008-15. 2. Burton TJ, Mackenzie IS, Balan K et al. Evaluation of the sensitivity and specificity of (11)C-metomidate positron emission tomography (PET)-CT for lateralizing aldosterone secretion by Conn’s adenomas. J Clin Endocrinol Metab 2012;97(1):100-9. 3. Hahner S, Kreissl MC, Fassnacht M et al. Functional characterization of adrenal lesions using [123I]IMTO-SPECT/CT. J Clin Endocrinol Metab 2013;98(4):1508-18. |
||||||||
|
Hyperplasie congénitale : freiner l’ACTH sans glucocorticoïdes ? Les antagonistes du récepteur du CRH de type 1 Le dilemme thérapeutique de l’hyperplasie congénitale par déficit en 21-hydroxylase est bien connu : freiner parfaitement l’ACTH (adréno-cortico-trophic-hormone) et la production d’androgènes expose au syndrome de Cushing, mais éviter le syndrome de Cushing en laissant filer l’ACTH… expose à l’excès d’androgènes. Trouver un juste milieu n’est pas facile et la réduction de la taille finale des enfants hyperplasiques, conséquence commune de ces deux écueils, en est la preuve. Le nœud du problème tient au fait que pour freiner le pic d’ACTH en fin de nuit on utilise des glucocorticoïdes par voie orale, donnés au plus tard au coucher : la surexposition aux glucocorticoïdes en début et milieu de nuit est inévitable et cette surexposition est néfaste sur le plan métabolique. Comment freiner le pic matinal d’ACTH sans amener trop de corticoïdes pendant la première partie de la nuit ? Jusqu’ici deux stratégies ont été développées : injecter l’hydrocortisone avec une pompe programmable comme on le fait avec l’insuline, ce qui donne des résultats intéressants mais au prix d’une contrainte assez lourde (1), ou mettre au point une forme galénique d’hydrocortisone qui permette sa libération retardée : approche qui a en fait déjà montré des résultats très prometteurs dans une étude de phase II (2). Dans un article très récent, l’équipe de Richard Auchus propose une nouvelle approche (3) : freiner l’ACTH, oui, mais sans glucocorticoïde, en utilisant des antagonistes du récepteur du CRH de type 1 (CRH-R1) . En somme, plutôt que d’appuyer lourdement sur le frein glucocorticoïde, levons le pied sur l’accélérateur CRH ! R. Auchus profite pour cela du fait que les antagonistes CRH-R1 sont actuellement développés par l’industrie pharmaceutique en raison de leurs potentiels anxiolytique et antidépresseur : les récepteurs CRH-R1 ne sont pas exprimés que dans l’hypophyse mais également dans le cortex cérébral où il seraient impliqués dans la réaction au stress. Il a déjà été démontré que ces antagonistes inhibent la sécrétion d’ACTH in vitro et in vivo et dans cet article R. Auchus teste un de ces antagonistes par voie orale, le NBI-77860, dans une étude de phase Ib, en simple aveugle. Dans cette étude huit patientes adultes porteuses d’une hyperplasie congénitale par déficit en 21-hydroxylase reçoivent le soir soit un placebo, soit une dose de 300 mg ou 600 mg de l’antagoniste, et l’objectif est de freiner l’ACTH et la 17-OH-progestérone dans la fenêtre de 8 à 12 h après l’administration vespérale de l’antagoniste (donc entre 6 et 10 h du matin). Les patientes ne prennent pas d’hydrocortisone pendant 20 h (de 6 h avant l’antagoniste à 14 h après). Les auteurs estiment qu’ils sont arrivés à cet objectif, avec une baisse (comparée au placebo) de l’ACTH et la 17-OH-progestérone qui est respectivement de 43 % et 0,7 % (!) pour la dose de 300 mg et 41 % et 27 % pour la dose de 600 mg. Les baisses d’androgènes sont encore plus modestes, mais les auteurs jugent cependant que la preuve de concept est faite et que cette étude pilote ouvre de nouvelles perspectives thérapeutiques. On ne demande qu’à l’espérer avec lui, cependant il faut souligner plusieurs limites de cette petite étude : – curieusement, il n’est précisé nulle part que les différences de taux d’ACTH et 17-OH-progestérone entre le placebo et l’antagoniste sont statistiquement significatives et on reste rêveur devant la baisse de 0,7 % de 17-OH-progestérone sous 300 mg d’antagoniste ; – si l’on admet le caractère significatif des différences, le fait que la baisse de 17-OH-progestérone soit moindre que celle d’ACTH suggère que la durée de freinage de l’ACTH est vraisemblablement trop courte pour freiner valablement la production de 17-OH-progestérone et a fortiori des androgènes ; – enfin, sur les 8 patientes R. Auchus précise que seules 4 sont répondeuses, en prenant comme critère de réponse une baisse de 17-OH-progestérone d’au moins 50 % sur un des points de la fenêtre. Au total, les antagonistes CRH-R1 sont une nouvelle approche qui représente un espoir pour les patients de freiner leur sécrétion d’androgènes sans les exposer au syndrome de Cushing, mais il reste encore du chemin à parcourir avant que ces antagonistes démontrent leur réelle efficacité. Il demeure également des questions sur des effets indésirables possibles : R. Auchus vante l’intérêt d’un freinage de l’ACTH sans glucocorticoïde mais il ne faut pas oublier que son frein n’agit pas que sur l’hypophyse : les antagonistes CRH-R1 agissent aussi sur le cortex cérébral, cible pour laquelle l’industrie pharmaceutique les développe. Les études à venir dans l’hyperplasie congénitale devront pouvoir enregistrer d’éventuels effets indésirables psychiques de ces composés mais on peut quand même se demander si le meilleur frein de l’ACTH ne reste pas l’hydrocortisone “au bon moment”, comme proposé dans par A. Mallapa (2). |
||||||||
|
Références bibliographiques 1. Hindmarsh PC. The child with difficult to control Congenital Adrenal Hyperplasia: is there a place for continuous subcutaneous hydrocortisone therapy. Clin endocrinol (Oxf) 2015;81(1):15-8. 2. Mallappa A, Sinaii N, Kumar P et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2015;100(3):1137-45. 3. Turcu AF, Spencer-Segal JL, Farber RH et al. Single-Dose Study of a Corticotropin-Releasing Factor Receptor-1 Antagonist in Women With 21-Hydroxylase Deficiency. J Clin Endocrinol Metab 2016;101(3):1174-80. |
||||||||
 |
||||||||
|
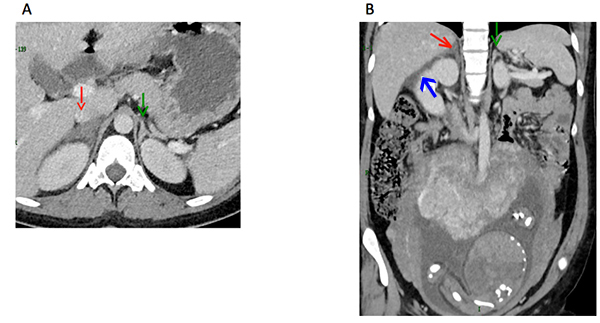
Ischémie surrénalienne droite révélée par une douleur aiguë du flanc D au 3e trimestre d’une grossesse Une patiente enceinte (32 semaines d’aménorrhée), se présente aux urgences pour une douleur aiguë du flanc D, évoquant une crise de colique néphrétique. L’échographie ne montre pas de dilatation des voies pyélocalicielles ni biliaires, il n’y a pas d’infection urinaire. Une exploration TDM (tomodensitométrie) est alors réalisée et met en évidence une ischémie surrénalienne droite (A) avec inflammation péritonéale en regard (B).
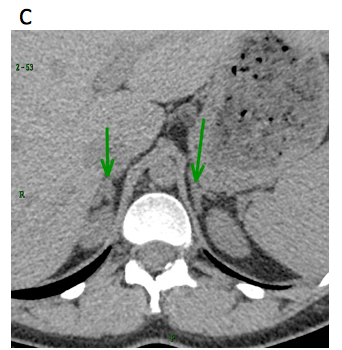
A. Coupe axiale 70 secondes après injection d’iode, montrant un élargissement et une absence de rehaussement de la surrénale droite (flèche rouge) sans nécrose et d’hématome. La surrénale gauche (flèche verte) se rehausse normalement. B. Coupe coronale 70 secondes après injection d’iode, montrant la tuméfaction surrénalienne droite (flèche rouge) et la lame liquidienne péritonéale droite (flèche bleue). La surrénale G a un réhaussement normal (flèche verte) utérus gravide. La patiente ne présente pas de signes d’insuffisance surrénalienne et garde un taux de cortisol plasmatique normal, l’évolution sous antalgique est simple. Après accouchement à terme, la patiente garde un test au synacthène normal et l’évaluation radiologique à 4 mois post-partum (sans injection) montre une surrénale droite d’aspect normal (C). C. Contrôle à 4 mois montrant une restitution ad integrum de la surrénale droite. Examen réalisé sans injection (la patiente l’a refusé). |
||||||||
|
1. Hoen N, Ziane G, Grange C, Raudrant D, Dupuis O 2011 [Unilateral adrenal ischemia during third trimester of pregnancy: about two cases]. Gynecologie, obstetrique & fertilite 39:e73-76. |
||||||||